DNA Methylation Biomarkers: Cancer and Beyond

Abstract
:
1. Introduction
2. Epigenetics and Disease Latency
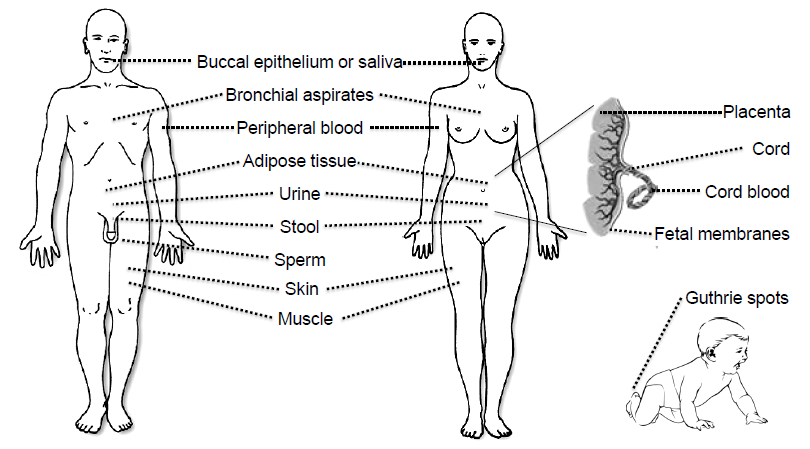
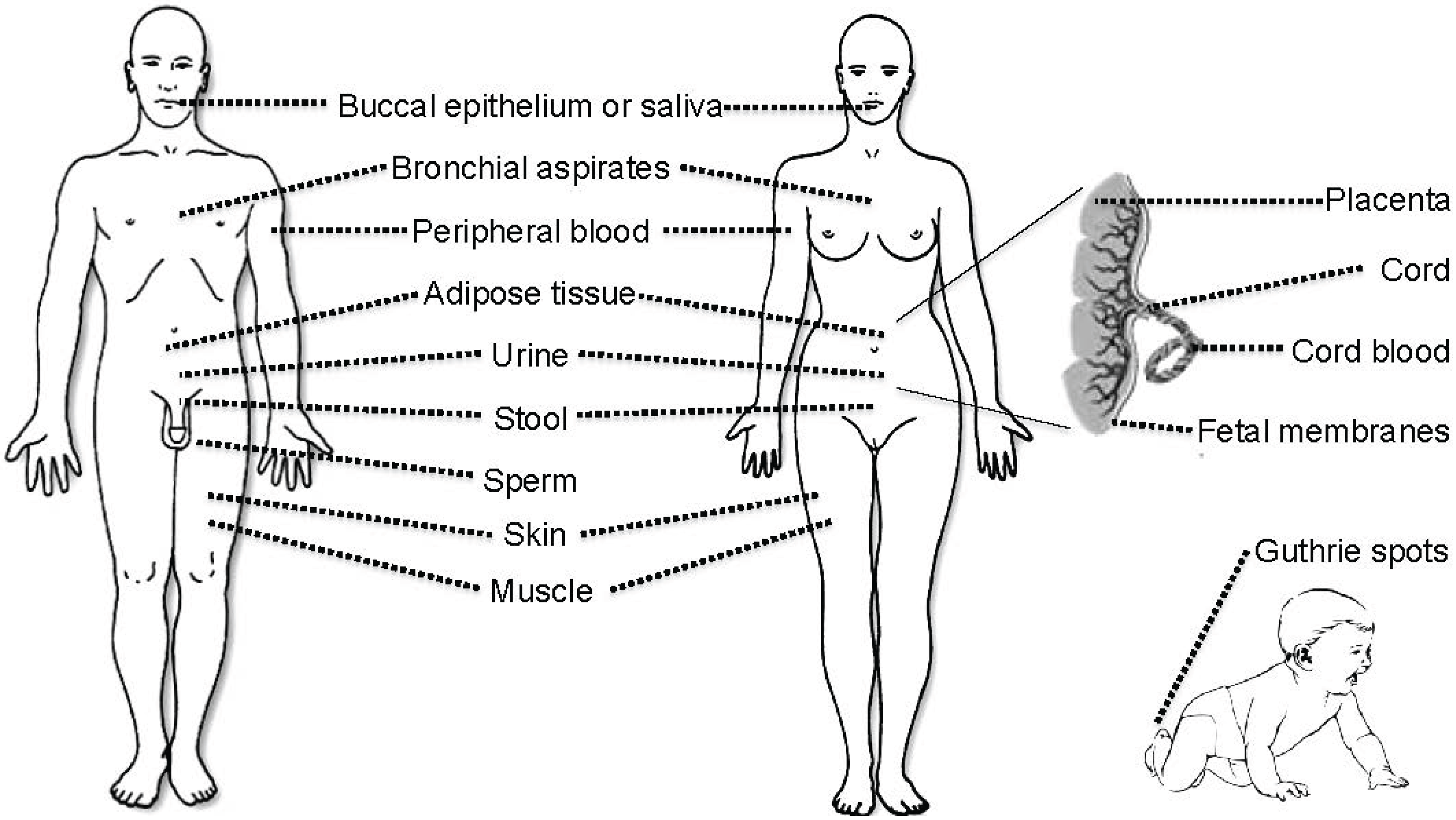
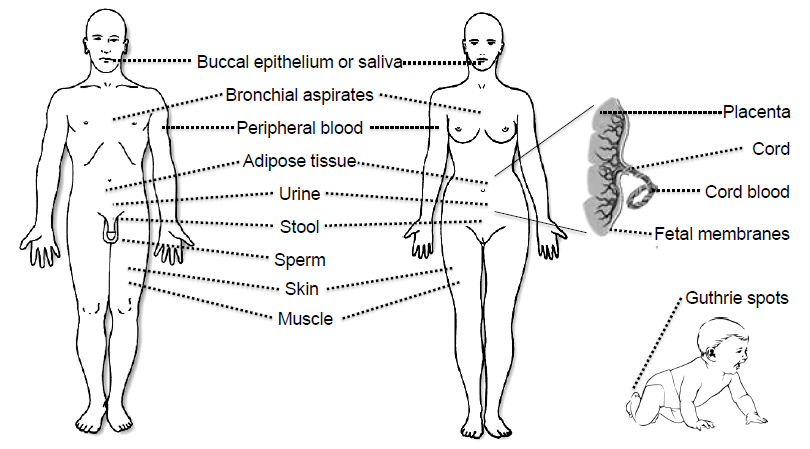
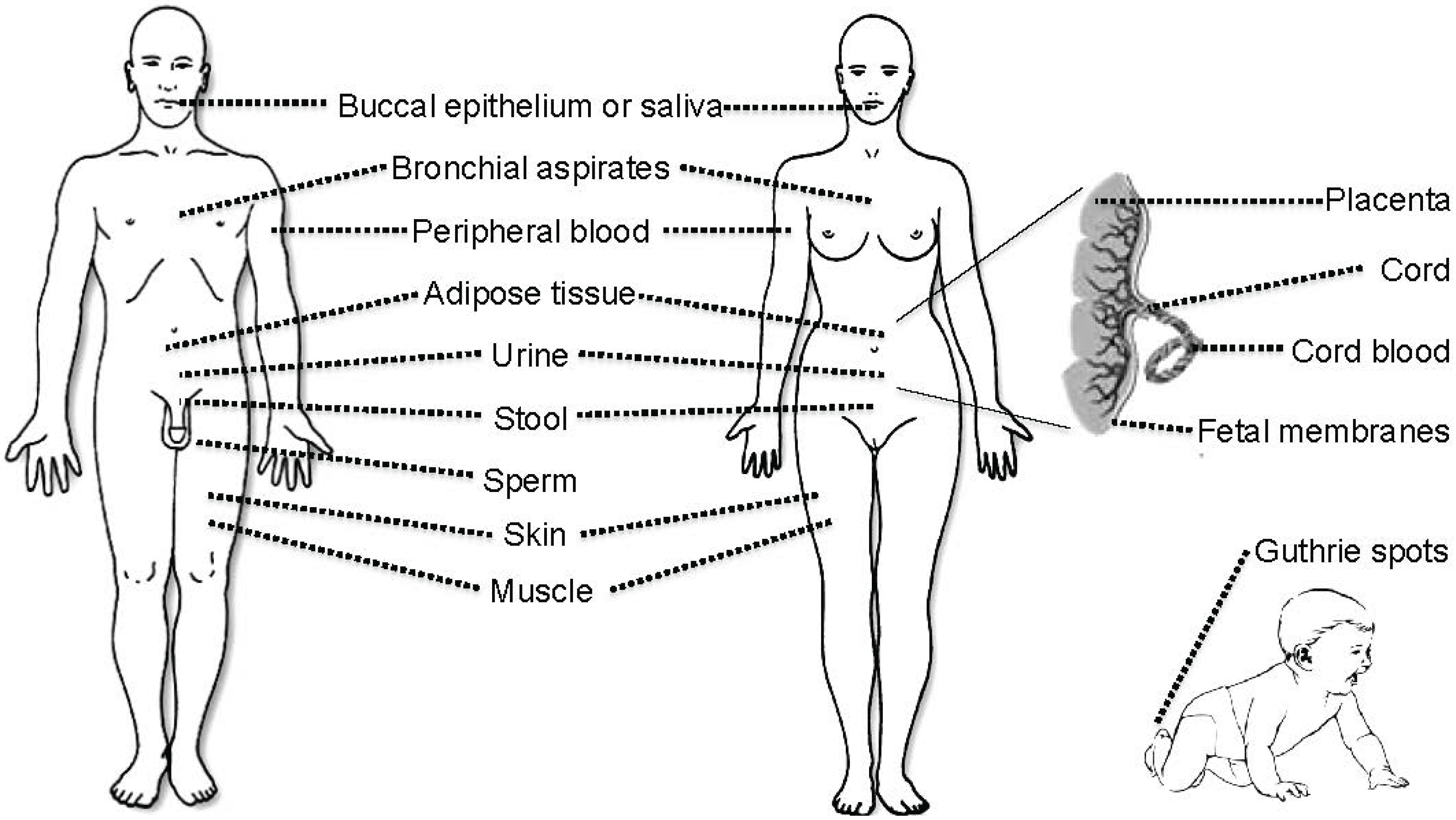
3. Tissues and Bodily Fluids Suitable for Analysis of DNA Methylation Biomarkers
4. Parameters for Developing DNA Methylation Biomarkers

{kind=link}
{kind=link}
| Nomenclature | Description |
|---|---|
| Potential biomarker | Results of a single study |
| Validated biomarker | Same finding using an independent method |
| Replicated biomarker | Same finding in independent cohort(s) |
| Candidate clinical biomarker | Replicated in multiple cohorts and subjected to systematic review and meta-analysis; most likely undergoing clinical trials |
| Proven clinical biomarker | Used in clinical practice |
4.1. Methods for DNA Methylation Biomarker Discovery
4.2. DNA Methylation Assay Sensitivity and Specificity
4.3. Barriers to Developing, Testing and Using DNA Methylation Biomarkers
5. Methods Suitable for the Analysis of Locus-Specific DNA Methylation Biomarkers
Lessons Learned from the DNA Methylation Biomarker MGMT
| Gene(s) | Type of Biomarker | Type of Cancer | Diagnostic Test Kit: Brand Name (Manufacturer) | References |
|---|---|---|---|---|
| VIM | diagnostic | Colorectal | Cologuard (Exact Sciences) | [128] 1 |
| SEPT9 | diagnostic | Colorectal | Epi proColon (Epigenomics) | [129] 1 |
| SHOX2 | diagnostic | Lung | Epi prolong (Epigenomics) | [130,131,132,133,134,135] 2 |
| GSTP1/APC/RASSF1A | diagnostic | Prostate | ConfirmMDx (MDx Health) | [136,137,138] 1 |
| MGMT | predictive | Glioblastoma | PredictMDx Glioblastoma (MDx Health) | [121,139,140] 1 |
6. DNA Methylation Biomarkers
6.1. Candidate Clinical DNA Methylation Biomarkers for Cancer
6.1.1. SHOX2
6.1.2. PITX2
6.1.3. MGMT
7. DNA Methylation Biomarkers for Genomic Imprinting Disorders
8. DNA Methylation Biomarkers of Outcome in Chronic Diseases Other than Cancer
8.1. DNA Methylation Biomarkers for Adverse Environments
8.1.1. AHRR Methylation and Smoking
| Reference | Platform | Age, Median | Exposure | Tissue | N | Effects Elsewhere in AHRR | AHRR Expression | Vali-dation | Repli-cation | Effect Size | Notes |
|---|---|---|---|---|---|---|---|---|---|---|---|
| [213] | HM450 | Adults, 45 | Current smoking | LCLs & alveolar MP | 119/19 1 | yes | Yes 2 | No | No | −15%/NS | |
| [209] | HM450 | Birth | Maternal smoking 3 | Whole CB | 1062/36 4 | Yes | No | No | Yes 4 | −7.5%/−7.7% 4 | Multiple hits in the aryl hydrocarbon signaling pathway. Authors have since shown that effects are specific for maternal smoking through at least gestational week 18 [218] |
| [207] | HM450 | Adults, 49 | Current smoking | PBMC | 111 | Yes | No | Yes | No | −15% | African Americans |
| [208] | HM450 | Adults, 48 | Current smoking | Whole PB | 81/84 5 | No | No | No | Yes 6 | −22% | Former smokers same as never smokers; changed only slightly after adjusting for cell composition |
| [210] | HM450 | Adults, 22 | Current serum cotinine | PBMC | 107 | yes | No | No | No | −20% 7 | |
| [211] | HM450 | Adults, 51/55/49/? 8 | Current smoking | Whole PB, lung tissue | 184/190/180/27 | yes | Yes 9 | Yes | Yes | −17%/−14%/NS/NS 10 | Replicated in a mouse model of smoking exposure |
| [212] | HM450 | Adults, 60/53 11 | Current smoking | Whole PB | 749/232 11 | yes | No | Yes | Yes 11 | −24/−23% 11 | methylation-specific protein binding patterns were observed for cg05575921; levels in former smokers revert to levels similar to never smokers over time |
| [215] | HM450 | Birth | Maternal smoking | Whole CB | 889 | yes | No | No | Yes | −4% | Replicated a previous study [209] |
| [214] | HM450 | Adults, 43 | Current smoking | Whole PB | 432 | yes | No 13 | No | Yes | −7.4% | Replicated a previous study [212]; no effect with tobacco snuff |
| [216] | HM450 | Female adults, 57 | Current smoking | Whole PB | 200 | No | No | No | Yes | −8% | Former and never smokers had similar methylation levels |
| [217] | Sequenom EpiTyper | Birth & 18 months | Maternal smoking | CBMC, buccal epithelium, placenta | 46/15/24 12 | yes | Yes 14 | n/a | Y | −10%/NS/NS 12 | No effect if mother smoked early pregnancy only; effects of smoking stable to 18 months of age |
| Probe | Gene | References |
|---|---|---|
| cg03991871 | AHRR | [209,212,213,215] |
| cg21161138 | AHRR | [207,209,210,211,212,215] |
| cg03636183 | F2RL3 | [208,211,214,216] |
| cg09935388 | GFI1 | [208,209,212,214,215] |
| cg22132788 | MYO1G | [208,209,210,214] |
| cg12803068 | MYO1G | [210,212,215,218] |
| cg21566642 | Intergenic (CpG island, DHS) | [207,208,211,212] |
| cg06126421 | Intergenic (enhancer, DHS) | [207,208,211,212,214] |
8.1.2. NR3C1 Methylation and Stress
8.2. DNA Methylation Risk Biomarkers at Birth
8.3. DNA Methylation Biomarkers during Childhood
8.4. DNA Methylation Biomarkers in Adults
8.5. DNA Methylation Biomarkers of Aging
9. Integrating Epigenetic Data into Disease Risk Models
10. Future Prospects
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Naylor, S. Biomarkers: Current perspectives and future prospects. Expert Rev. Mol. Diagn. 2003, 3, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R. Biomarkers: Potential uses and limitations. NeuroRx 2004, 1, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Strimbu, K.; Tavel, J.A. What are biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Lochhead, P.; Chan, A.T.; Nishihara, R.; Cho, E.; Wolpin, B.M.; Meyerhardt, J.A.; Meissner, A.; Schernhammer, E.S.; Fuchs, C.S.; et al. Molecular pathological epidemiology of epigenetics: Emerging integrative science to analyze environment, host, and disease. Mod. Pathol. 2013, 26, 465–484. [Google Scholar]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Teh, A.L.; Pan, H.; Chen, L.; Ong, M.L.; Dogra, S.; Wong, J.; Macisaac, J.L.; Mah, S.M.; McEwen, L.M.; Saw, S.M.; et al. The effect of genotype and in utero environment on inter-individual variation in neonate DNA methylomes. Genome Res. 2014. [Google Scholar] [CrossRef]
- De Laat, W.; Duboule, D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature 2013, 502, 499–506. [Google Scholar]
- Quina, A.S.; Buschbeck, M.; di Croce, L. Chromatin structure and epigenetics. Biochem. Pharmacol. 2006, 72, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and function of mammalian DNA methyltransferases. Chembiochem 2011, 12, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Jurkowska, R.Z. New concepts in DNA methylation. Trends Biochem. Sci. 2014, 39, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar]
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Han, L. CpG islands: Algorithms and applications in methylation studies. Biochem. Biophys. Res. Commun. 2009, 382, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Caffo, B.; Jaffee, H.A.; Irizarry, R.A.; Feinberg, A.P. Redefining CpG islands using hidden Markov models. Biostatistics 2010, 11, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Chuang, L.Y.; Huang, H.C.; Lin, M.C.; Yang, C.H. Particle swarm optimization with reinforcement learning for the prediction of CpG islands in the human genome. PLoS One 2011, 6, e21036. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Queiros, A.C.; Beekman, R.; Martin-Subero, J.I. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim. Biophys. Acta 2013, 1829, 1161–1174. [Google Scholar] [CrossRef]
- Oberdoerffer, S. A conserved role for intragenic DNA methylation in alternative pre-mRNA splicing. Transcription 2012, 3, 106–109. [Google Scholar] [PubMed]
- Illingworth, R.S.; Gruenewald-Schneider, U.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Smith, C.; Harrison, D.J.; Andrews, R.; Bird, A.P. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010, 6, e1001134. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA methylation and cancer-associated genetic instability. Adv. Exp. Med. Biol. 2005, 570, 363–392. [Google Scholar] [PubMed]
- Choi, J.D.; Lee, J.S. Interplay between Epigenetics and Genetics in Cancer. Genomics Inform. 2013, 11, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Liu, G.; Stevens, J.B.; Bremer, S.W.; Ye, K.J.; Ye, C.J. Genetic and epigenetic heterogeneity in cancer: The ultimate challenge for drug therapy. Curr. Drug Targets 2010, 11, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. Genetic and non-genetic instability in tumor progression: Link between the fitness landscape and the epigenetic landscape of cancer cells. Cancer Metastasis Rev. 2013, 32, 423–448. [Google Scholar] [CrossRef] [PubMed]
- Stecklein, S.R.; Jensen, R.A.; Pal, A. Genetic and epigenetic signatures of breast cancer subtypes. Front. Biosci. 2012, 4, 934–949. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Buklijas, T. A conceptual framework for the developmental origins of health and disease. J. Dev. Orig. Health Dis. 2010, 1, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A.; Pinal, C. The developmental origins of adult disease. Matern. Child Nutr. 2005, 1, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The fetal and infant origins of adult disease. BMJ 1990, 301, 1111. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.M.; Lillycrop, K.A.; Burdge, G.C.; Gluckman, P.D.; Hanson, M.A. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr. Res. 2007, 61, R5–R10. [Google Scholar]
- McNeal, C.J.; Wilson, D.P.; Christou, D.; Bush, R.L.; Shepherd, L.G.; Santiago, J.; Wu, G.Y. The use of surrogate vascular markers in youth at risk for premature cardiovascular disease. J. Pediatr. Endocrinol. Metab. 2009, 22, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Urbina, E.M.; Williams, R.V.; Alpert, B.S.; Collins, R.T.; Daniels, S.R.; Hayman, L.; Jacobson, M.; Mahoney, L.; Mietus-Snyder, M.; Rocchini, A.; et al. Noninvasive assessment of subclinical atherosclerosis in children and adolescents: Recommendations for standard assessment for clinical research: A scientific statement from the American Heart Association. Hypertension 2009, 54, 919–950. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.C.; Morley, R.; Saffery, R.; Craig, J.M. Archived guthrie blood spots as a novel source for quantitative DNA methylation analysis. Biotechniques 2008, 45, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Thirlwell, C.; Eymard, M.; Feber, A.; Teschendorff, A.; Pearce, K.; Lechner, M.; Widschwendter, M.; Beck, S. Genome-wide DNA methylation analysis of archival formalin-fixed paraffin-embedded tissue using the Illumina Infinium HumanMethylation27 BeadChip. Methods 2010, 52, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.C.; Ashley, D.; Chatterton, Z.; Parkinson-Bates, M.; Ng, H.K.; Halemba, M.S.; Kowalczyk, A.; Bedo, J.; Wang, Q.; Bell, K.; et al. A distinct DNA methylation signature defines pediatric pre-B cell acute lymphoblastic leukemia. Epigenetics 2012, 7, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Ribel-Madsen, R.; Fraga, M.F.; Jacobsen, S.; Bork-Jensen, J.; Lara, E.; Calvanese, V.; Fernandez, A.F.; Friedrichsen, M.; Vind, B.F.; Hojlund, K.; et al. Genome-wide analysis of DNA methylation differences in muscle and fat from monozygotic twins discordant for type 2 diabetes. PLoS One 2012, 7, e51302. [Google Scholar]
- Talens, R.P.; Boomsma, D.I.; Tobi, E.W.; Kremer, D.; Jukema, J.W.; Willemsen, G.; Putter, H.; Slagboom, P.E.; Heijmans, B.T. Variation, patterns, and temporal stability of DNA methylation: Considerations for epigenetic epidemiology. FASEB J. 2010, 24, 3135–3144. [Google Scholar] [CrossRef] [PubMed]
- Souren, N.Y.; Tierling, S.; Fryns, J.P.; Derom, C.; Walter, J.; Zeegers, M.P. DNA methylation variability at growth-related imprints does not contribute to overweight in monozygotic twins discordant for BMI. Obesity 2011, 19, 1519–1522. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, S.A.; Palmisano, W.A.; Gilliland, F.D.; Crooks, L.A.; Divine, K.K.; Winters, S.A.; Grimes, M.J.; Harms, H.J.; Tellez, C.S.; Smith, T.M.; et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 2002, 62, 2370–2377. [Google Scholar] [PubMed]
- Barnes, S.K.; Ozanne, S.E. Pathways linking the early environment to long-term health and lifespan. Prog. Biophys. Mol. Biol. 2011, 106, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Ollikainen, M.; Smith, K.R.; Joo, E.J.; Ng, H.K.; Andronikos, R.; Novakovic, B.; Abdul Aziz, N.K.; Carlin, J.B.; Morley, R.; Saffery, R.; et al. DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome. Hum. Mol. Genet. 2010, 19, 4176–4188. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, B.; Buiting, K.; Gross, S.; Horsthemke, B. Characterization of a methylation imprint in the Prader-Willi syndrome chromosome region. Hum. Mol. Genet. 1993, 2, 1995–1999. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.M.; Sheppard, A.; Gluckman, P.D.; Lillycrop, K.A.; Burdge, G.C.; McLean, C.; Rodford, J.; Slater-Jefferies, J.L.; Garratt, E.; Crozier, S.R.; et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes 2011, 60, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Rakyan, V.K. Correcting for cell-type composition bias in epigenome-wide association studies. Genome Med. 2014, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Adalsteinsson, B.T.; Gudnason, H.; Aspelund, T.; Harris, T.B.; Launer, L.J.; Eiriksdottir, G.; Smith, A.V.; Gudnason, V. Heterogeneity in white blood cells has potential to confound DNA methylation measurements. PLoS One 2012, 7, e46705. [Google Scholar] [CrossRef] [PubMed]
- Loh, M.; Liem, N.; Lim, P.L.; Vaithilingam, A.; Cheng, C.L.; Salto-Tellez, M.; Yong, W.P.; Soong, R. Impact of sample heterogeneity on methylation analysis. Diagn. Mol. Pathol. 2010, 19, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Montano, C.M.; Irizarry, R.A.; Kaufmann, W.E.; Talbot, K.; Gur, R.E.; Feinberg, A.P.; Taub, M.A. Measuring cell-type specific differential methylation in human brain tissue. Genome Biol. 2013, 14, R94. [Google Scholar] [CrossRef] [PubMed]
- Moverare-Skrtic, S.; Mellstrom, D.; Vandenput, L.; Ehrich, M.; Ohlsson, C. Peripheral blood leukocyte distribution and body mass index are associated with the methylation pattern of the androgen receptor promoter. Endocrine 2009, 35, 204–210. [Google Scholar]
- Zou, J.; Lippert, C.; Heckerman, D.; Aryee, M.; Listgarten, J. Epigenome-wide association studies without the need for cell-type composition. Nat. Methods 2014, 11, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Accomando, W.P.; Wiencke, J.K.; Houseman, E.A.; Nelson, H.H.; Kelsey, K.T. Quantitative reconstruction of leukocyte subsets using DNA methylation. Genome Biol. 2014, 15, R50. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef]
- Herceg, Z.; Hainaut, P. Genetic and epigenetic alterations as biomarkers for cancer detection, diagnosis and prognosis. Mol. Oncol. 2007, 1, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Guler, G.; Han, S.Y.; Johnston, D.; Druck, T.; McCorkell, K.A.; Palazzo, J.; McCue, P.A.; Baffa, R.; Huebner, K. Fragile genes as biomarkers: Epigenetic control of WWOX and FHIT in lung, breast and bladder cancer. Oncogene 2005, 24, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Manne, U. Genetic and epigenetic biomarkers in cancer diagnosis and identifying high risk populations. Crit. Rev. Oncol./Hematol. 2006, 60, 9–18. [Google Scholar]
- Zilberman, D.; Henikoff, S. Genome-wide analysis of DNA methylation patterns. Development 2007, 134, 3959–3965. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Tycko, B.; Liu, T.M.; Lin, J.J.; Huang, T.H. Methods in DNA methylation profiling. Epigenomics 2009, 1, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Kalari, S.; Pfeifer, G.P. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv. Genet. 2010, 70, 277–308. [Google Scholar] [PubMed]
- Kim, M.; Long, T.I.; Arakawa, K.; Wang, R.; Yu, M.C.; Laird, P.W. DNA methylation as a biomarker for cardiovascular disease risk. PLoS One 2010, 5, e9692. [Google Scholar] [CrossRef] [PubMed]
- How Kit, A.; Nielsen, H.M.; Tost, J. DNA methylation based biomarkers: Practical considerations and applications. Biochimie 2012, 94, 2314–2337. [Google Scholar]
- Michels, K.B.; Binder, A.M.; Dedeurwaerder, S.; Epstein, C.B.; Greally, J.M.; Gut, I.; Houseman, E.A.; Izzi, B.; Kelsey, K.T.; Meissner, A.; et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat. Methods 2013, 10, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011, 12, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Wians, F.H. Clinical laboratory tests: Which, why, and what do the results mean? Lab Med. 2009, 40, 105–113. [Google Scholar] [CrossRef]
- Linnet, K.; Bossuyt, P.M.; Moons, K.G.; Reitsma, J.B. Quantifying the accuracy of a diagnostic test or marker. Clin. Chem. 2012, 58, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Wacholder, S. From differences in means between cases and controls to risk stratification: A business plan for biomarker development. Cancer Discov. 2013, 3, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Esteller, M. DNA methylation: A profile of methods and applications. Biotechniques 2002, 33, 632, 634, 636–649. [Google Scholar]
- Dahl, C.; Guldberg, P. DNA methylation analysis techniques. Biogerontology 2003, 4, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Hansen, L.L. PCR-based methods for detecting single-locus DNA methylation biomarkers in cancer diagnostics, prognostics, and response to treatment. Clin. Chem. 2009, 55, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, H.G.; Tse, M.Y.; Pang, S.C.; Arboleda, H.; Forero, D.A. Optimizing methodologies for PCR-based DNA methylation analysis. Biotechniques 2013, 55, 181–197. [Google Scholar] [PubMed]
- Jorda, M.; Peinado, M.A. Methods for DNA methylation analysis and applications in colon cancer. Mutat. Res. 2010, 693, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Waterland, R.A. Methods of DNA methylation analysis. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Grunau, C.; Clark, S.J.; Rosenthal, A. Bisulfite genomic sequencing: Systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001, 29, e65. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.E.; Jung, M.; Meller, S.; Leisse, A.; Sailer, V.; Zech, J.; Mengdehl, M.; Garbe, L.A.; Uhl, B.; Kristiansen, G.; et al. Performance evaluation of kits for bisulfite-conversion of DNA from tissues, cell lines, FFPE tissues, aspirates, lavages, effusions, plasma, serum, and urine. PLoS One 2014, 9, e93933. [Google Scholar] [CrossRef]
- Tanaka, K.; Okamoto, A. Degradation of DNA by bisulfite treatment. Bioorg. Med. Chem. Lett. 2007, 17, 1912–1915. [Google Scholar] [CrossRef] [PubMed]
- Munson, K.; Clark, J.; Lamparska-Kupsik, K.; Smith, S.S. Recovery of bisulfite-converted genomic sequences in the methylation-sensitive QPCR. Nucleic Acids Res. 2007, 35, 2893–2903. [Google Scholar] [CrossRef] [PubMed]
- Brandes, J.C.; Carraway, H.; Herman, J.G. Optimal primer design using the novel primer design program: MSPprimer provides accurate methylation analysis of the ATM promoter. Oncogene 2007, 26, 6229–6237. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, P.M.; Stirzaker, C.; Melki, J.R.; Millar, D.S.; Paul, C.L.; Clark, S.J. Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite-treated DNA. Nucleic Acids Res. 1997, 25, 4422–4426. [Google Scholar] [CrossRef] [PubMed]
- Chhibber, A.; Schroeder, B.G. Single-molecule polymerase chain reaction reduces bias: Application to DNA methylation analysis by bisulfite sequencing. Anal. Biochem. 2008, 377, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Moskalev, E.A.; Zavgorodnij, M.G.; Majorova, S.P.; Vorobjev, I.A.; Jandaghi, P.; Bure, I.V.; Hoheisel, J.D. Correction of PCR-bias in quantitative DNA methylation studies by means of cubic polynomial regression. Nucleic Acids Res. 2011, 39, e77. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Guo, Y.; Chen, X.; Ahmed, S.; Issa, J.P. Optimizing annealing temperature overcomes bias in bisulfite pcr methylation analysis. Biotechniques 2007, 42, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Wojdacz, T.K.; Hansen, L.L.; Dobrovic, A. A new approach to primer design for the control of PCR bias in methylation studies. BMC Res. Notes 2008, 1, 54. [Google Scholar] [CrossRef] [PubMed]
- Claus, R.; Wilop, S.; Hielscher, T.; Sonnet, M.; Dahl, E.; Galm, O.; Jost, E.; Plass, C. A systematic comparison of quantitative high-resolution DNA methylation analysis and methylation-specific PCR. Epigenetics 2012, 7, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.; Tanner, S.M.; Byrd, J.C.; Angerman, E.B.; Perko, J.D.; Chen, S.S.; Hackanson, B.; Grever, M.R.; Lucas, D.M.; Matkovic, J.J.; et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 2007, 129, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Lin, B.; Sibenaller, Z.; Ryken, T.; Lee, H.; Yoon, J.G.; Rostad, S.; Foltz, G. Comprehensive analysis of MGMT promoter methylation: Correlation with MGMT expression and clinical response in GBM. PLoS One 2011, 6, e16146. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Candiloro, I.L.; Dobrovic, A. The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics 2010, 2, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.N.; Jiang, W.W.; Kim, M.; Benoit, N.; Taylor, R.; Clinger, J.; Sidransky, D.; Califano, J.A. Death-associated protein kinase promoter hypermethylation in normal human lymphocytes. Cancer Res. 2003, 63, 7694–7698. [Google Scholar] [PubMed]
- Srinivasan, M.; Sedmak, D.; Jewell, S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am. J. Pathol. 2002, 161, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Colella, S.; Shen, L.; Baggerly, K.A.; Issa, J.P.; Krahe, R. Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. Biotechniques 2003, 35, 146–150. [Google Scholar] [PubMed]
- Tost, J.; Dunker, J.; Gut, I.G. Analysis and quantification of multiple methylation variable positions in CpG islands by Pyrosequencing. Biotechniques 2003, 35, 152–156. [Google Scholar] [PubMed]
- Mikeska, T.; Felsberg, J.; Hewitt, C.A.; Dobrovic, A. Analysing DNA methylation using bisulphite pyrosequencing. Methods Mol. Biol. 2011, 791, 33–53. [Google Scholar] [PubMed]
- Dejeux, E.; Audard, V.; Cavard, C.; Gut, I.G.; Terris, B.; Tost, J. Rapid identification of promoter hypermethylation in hepatocellular carcinoma by pyrosequencing of etiologically homogeneous sample pools. J. Mol. Diagn. 2007, 9, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Phillips, E.S.; Torrens, C.; Hanson, M.A.; Jackson, A.A.; Burdge, G.C. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR alpha promoter of the offspring. Br. J. Nutr. 2008, 100, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Ehrich, M.; Nelson, M.R.; Stanssens, P.; Zabeau, M.; Liloglou, T.; Xinarianos, G.; Cantor, C.R.; Field, J.K.; van den Boom, D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc. Natl. Acad. Sci. USA 2005, 102, 15785–15790. [Google Scholar] [CrossRef] [PubMed]
- Coolen, M.W.; Statham, A.L.; Gardiner-Garden, M.; Clark, S.J. Genomic profiling of CpG methylation and allelic specificity using quantitative high-throughput mass spectrometry: Critical evaluation and improvements. Nucleic Acids Res. 2007, 35, e119. [Google Scholar] [CrossRef] [PubMed]
- Wojdacz, T.K.; Dobrovic, A. Methylation-sensitive high resolution melting (MS-HRM): A new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 2007, 35, e41. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Dobrovic, A. Methylation-sensitive high resolution melting for the rapid analysis of DNA methylation. In Epigenetics: A Reference Manual; Craig, J.M., Wong, N.C., Eds.; Caister Academic Press: Norwich, UK, 2011; pp. 325–335. [Google Scholar]
- Candiloro, I.L.; Mikeska, T.; Dobrovic, A. Closed-tube PCR methods for locus-specific DNA methylation analysis. Methods Mol. Biol. 2011, 791, 55–71. [Google Scholar] [PubMed]
- Candiloro, I.L.; Mikeska, T.; Dobrovic, A. Assessing combined methylation-sensitive high resolution melting and pyrosequencing for the analysis of heterogeneous DNA methylation. Epigenetics 2011, 6, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Elezi, L.; Hainfellner, J.A. Reliability and reproducibility of PCR-based testing of O6-methylguanine-DNA methyltransferase gene (MGMT) promoter methylation status in formalin-fixed and paraffin-embedded neurosurgical biopsy specimens. Clin. Neuropathol. 2008, 27, 388–390. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.G.; Roldan, G.; Magliocco, A.; McIntyre, J.B.; Parney, I.; Easaw, J.C. Determination of the methylation status of MGMT in different regions within glioblastoma multiforme. J. Neurooncol. 2011, 102, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, e32. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.; Qu, W.; Ho, T.; Clark, S.J.; Molloy, P. Conversion-specific detection of DNA methylation using real-time polymerase chain reaction (ConLight-MSP) to avoid false positives. Methods 2002, 27, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, C.; Shehi, E.; Adlerstein, D.; Makrigiorgos, G.M. MS-FLAG, a novel real-time signal generation method for methylation-specific PCR. Clin. Chem. 2007, 53, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Mikeska, T.; Krypuy, M.; Dobrovic, A. Sensitive Melting Analysis after Real Time- Methylation Specific PCR (SMART-MSP): High-throughput and probe-free quantitative DNA methylation detection. Nucleic Acids Res. 2008, 36, e42. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, S.E.; Distler, J.; Goodman, N.S.; Mooney, S.H.; Kluth, A.; Olek, A.; Schwope, I.; Tetzner, R.; Ziebarth, H.; Berlin, K. A real-time PCR assay for DNA-methylation using methylation-specific blockers. Nucleic Acids Res. 2004, 32, e10. [Google Scholar] [CrossRef] [PubMed]
- Nygren, A.O.; Ameziane, N.; Duarte, H.M.; Vijzelaar, R.N.; Waisfisz, Q.; Hess, C.J.; Schouten, J.P.; Errami, A. Methylation-specific MLPA (MS-MLPA): Simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res. 2005, 33, e128. [Google Scholar] [CrossRef] [PubMed]
- Serizawa, R.R.; Ralfkiaer, U.; Dahl, C.; Lam, G.W.; Hansen, A.B.; Steven, K.; Horn, T.; Guldberg, P. Custom-designed MLPA using multiple short synthetic probes: Application to methylation analysis of five promoter CpG islands in tumor and urine specimens from patients with bladder cancer. J. Mol. Diagn. 2010, 12, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Gatta, V.; Gennaro, E.; Franchi, S.; Cecconi, M.; Antonucci, I.; Tommasi, M.; Palka, G.; Coviello, D.; Stuppia, L.; Grasso, M. MS-MLPA analysis for FMR1 gene: Evaluation in a routine diagnostic setting. BMC Med. Genet. 2013, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Jeuken, J.W.; Cornelissen, S.J.; Vriezen, M.; Dekkers, M.M.; Errami, A.; Sijben, A.; Boots-Sprenger, S.H.; Wesseling, P. MS-MLPA: An attractive alternative laboratory assay for robust, reliable, and semiquantitative detection of MGMT promoter hypermethylation in gliomas. Lab. Investig. 2007, 87, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Homig-Holzel, C.; Savola, S. Multiplex ligation-dependent probe amplification (MLPA) in tumor diagnostics and prognostics. Diagn. Mol. Pathol. 2012, 21, 189–206. [Google Scholar]
- Tano, K.; Shiota, S.; Collier, J.; Foote, R.S.; Mitra, S. Isolation and structural characterization of a cDNA clone encoding the human DNA repair protein for O6-alkylguanine. Proc. Natl. Acad. Sci. USA 1990, 87, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.T.; Vermeulen, S.; Darroudi, F.; Valentine, M.B.; Brent, T.P.; Mitra, S.; Tano, K. Chromosomal localization of human O6-methylguanine-DNA methyltransferase (MGMT) gene by in situ hybridization. Mutagenesis 1992, 7, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Nutt, C.L.; Costello, J.F.; Bambrick, L.L.; Yarosh, D.B.; Swinnen, L.J.; Chambers, A.F.; Cairncross, J.G. O6-methylguanine-DNA methyltransferase in tumors and cells of the oligodendrocyte lineage. Can. J. Neurol. Sci. 1995, 22, 111–115. [Google Scholar] [PubMed]
- Mineura, K.; Yanagisawa, T.; Watanabe, K.; Kowada, M.; Yasui, N. Human brain tumor O(6)-methylguanine-DNA methyltransferase mRNA and its significance as an indicator of selective chloroethylnitrosourea chemotherapy. Int. J. Cancer 1996, 69, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Silber, J.R.; Bobola, M.S.; Ghatan, S.; Blank, A.; Kolstoe, D.D.; Berger, M.S. O6-methylguanine-DNA methyltransferase activity in adult gliomas: Relation to patient and tumor characteristics. Cancer Res. 1998, 58, 1068–1073. [Google Scholar] [PubMed]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Godard, S.; Dietrich, P.Y.; Regli, L.; Ostermann, S.; Otten, P.; van Melle, G.; de Tribolet, N.; Stupp, R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004, 10, 1871–1874. [Google Scholar] [CrossRef] [PubMed]
- Paz, M.F.; Yaya-Tur, R.; Rojas-Marcos, I.; Reynes, G.; Pollan, M.; Aguirre-Cruz, L.; Garcia-Lopez, J.L.; Piquer, J.; Safont, M.J.; Balana, C.; et al. CpG island hypermethylation of the DNA repair enzyme methyltransferase predicts response to temozolomide in primary gliomas. Clin. Cancer Res. 2004, 10, 4933–4938. [Google Scholar]
- Blanc, J.L.; Wager, M.; Guilhot, J.; Kusy, S.; Bataille, B.; Chantereau, T.; Lapierre, F.; Larsen, C.J.; Karayan-Tapon, L. Correlation of clinical features and methylation status of MGMT gene promoter in glioblastomas. J. Neurooncol. 2004, 68, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar]
- Wick, W.; Weller, M.; van den Bent, M.; Sanson, M.; Weiler, M.; von Deimling, A.; Plass, C.; Hegi, M.; Platten, M.; Reifenberger, G. MGMT testing-the challenges for biomarker-based glioma treatment. Nat. Rev. Neurol. 2014, 10, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Christians, A.; Hartmann, C.; Benner, A.; Meyer, J.; von Deimling, A.; Weller, M.; Wick, W.; Weiler, M. Prognostic value of three different methods of MGMT promoter methylation analysis in a prospective trial on newly diagnosed glioblastoma. PLoS One 2012, 7, e33449. [Google Scholar] [CrossRef] [PubMed]
- Karayan-Tapon, L.; Quillien, V.; Guilhot, J.; Wager, M.; Fromont, G.; Saikali, S.; Etcheverry, A.; Hamlat, A.; Loussouarn, D.; Campion, L.; et al. Prognostic value of O6-methylguanine-DNA methyltransferase status in glioblastoma patients, assessed by five different methods. J. Neurooncol. 2010, 97, 311–322. [Google Scholar]
- Mikeska, T.; Bock, C.; el-Maarri, O.; Hubner, A.; Ehrentraut, D.; Schramm, J.; Felsberg, J.; Kahl, P.; Buttner, R.; Pietsch, T.; et al. Optimization of quantitative MGMT promoter methylation analysis using pyrosequencing and combined bisulfite restriction analysis. J. Mol. Diagn. 2007, 9, 368–381. [Google Scholar]
- Quillien, V.; Lavenu, A.; Karayan-Tapon, L.; Carpentier, C.; Labussiere, M.; Lesimple, T.; Chinot, O.; Wager, M.; Honnorat, J.; Saikali, S.; et al. Comparative assessment of 5 methods (methylation-specific polymerase chain reaction, MethyLight, pyrosequencing, methylation-sensitive high-resolution melting, and immunohistochemistry) to analyze O6-methylguanine-DNA-methyltranferase in a series of 100 glioblastoma patients. Cancer 2012, 118, 4201–4211. [Google Scholar]
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; van den Bent, M.J.; Wick, W.; Hegi, M.E. MGMT promoter methylation in malignant gliomas: Ready for personalized medicine? Nat. Rev. Neurol. 2010, 6, 39–51. [Google Scholar] [CrossRef]
- Mikeska, T.; Bock, C.; Do, H.; Dobrovic, A. DNA methylation biomarkers in cancer: Progress towards clinical implementation. Expert Rev. Mol. Diagn. 2012, 12, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.W.; Kong, F.M.; Zhou, J.P.; Dong, M. Aberrant promoter methylation of the vimentin gene may contribute to colorectal carcinogenesis: A meta-analysis. Tumour Biol. 2014, 35, 6783–6790. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.R. From discovery to the clinic: The novel DNA methylation biomarker (m)sept9 for the detection of colorectal cancer in blood. Epigenomics 2010, 2, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Darwiche, K.; Zarogoulidis, P.; Baehner, K.; Welter, S.; Tetzner, R.; Wohlschlaeger, J.; Theegarten, D.; Nakajima, T.; Freitag, L. Assessment of shox2 methylation in ebus-tbna specimen improves accuracy in lung cancer staging. Ann. Oncol. 2013, 24, 2866–2870. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, D.; Hasinger, O.; Liebenberg, V.; Field, J.K.; Kristiansen, G.; Soltermann, A. DNA methylation of the homeobox genes pitx2 and shox2 predicts outcome in non-small-cell lung cancer patients. Diagn. Mol. Pathol. 2012, 21, 93–104. [Google Scholar] [PubMed]
- Dietrich, D.; Jung, M.; Puetzer, S.; Leisse, A.; Holmes, E.E.; Meller, S.; Uhl, B.; Schatz, P.; Ivascu, C.; Kristiansen, G. Diagnostic and prognostic value of shox2 and sept9 DNA methylation and cytology in benign, paramalignant and malignant pleural effusions. PloS One 2013, 8, e84225. [Google Scholar] [CrossRef] [PubMed]
- Ilse, P.; Biesterfeld, S.; Pomjanski, N.; Fink, C.; Schramm, M. Shox2 DNA methylation is a tumour marker in pleural effusions. Cancer genomics proteomics 2013, 10, 217–223. [Google Scholar] [PubMed]
- Kneip, C.; Schmidt, B.; Seegebarth, A.; Weickmann, S.; Fleischhacker, M.; Liebenberg, V.; Field, J.K.; Dietrich, D. Shox2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. J. Thorac. Oncol. 2011, 6, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.; Liebenberg, V.; Dietrich, D.; Schlegel, T.; Kneip, C.; Seegebarth, A.; Flemming, N.; Seemann, S.; Distler, J.; Lewin, J.; et al. Shox2 DNA methylation is a biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer 2010, 10, 600. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Giovannucci, E.; Welge, J.; Mallick, P.; Tang, W.Y.; Ho, S.M. Measurement of gstp1 promoter methylation in body fluids may complement psa screening: A meta-analysis. Br. J. Cancer 2011, 105, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, J.; Yu, X.; Li, S.; Zhang, X.; Mo, Z.; Hu, Y. Apc gene hypermethylation and prostate cancer: A systematic review and meta-analysis. Eur. J. Hum. Genet. 2013, 21, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Chen, J.; Zhang, B.; Chen, X.; Huang, B.; Zhuang, J.; Mo, C.; Qiu, S. Association between rassf1a promoter methylation and prostate cancer: A systematic review and meta-analysis. PloS One 2013, 8, e75283. [Google Scholar] [PubMed]
- Yin, A.A.; Zhang, L.H.; Cheng, J.X.; Dong, Y.; Liu, B.L.; Han, N.; Zhang, X. The predictive but not prognostic value of mgmt promoter methylation status in elderly glioblastoma patients: A meta-analysis. PloS One 2014, 9, e85102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, X.Q.; Zhou, B.; Zhang, L. The prognostic value of mgmt promoter methylation in glioblastoma multiforme: A meta-analysis. Fam. cancer 2013, 12, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Berghoff, A.S.; Manzl, C.; Filipits, M.; Weinhausel, A.; Pulverer, W.; Dieckmann, K.; Widhalm, G.; Wohrer, A.; Knosp, E.; et al. Clinical neuropathology practice news 1–2014: Pyrosequencing meets clinical and analytical performance criteria for routine testing of mgmt promoter methylation status in glioblastoma. Clin. Neuropathol. 2014, 33, 6–14. [Google Scholar]
- Quillien, V.; Lavenu, A.; Sanson, M.; Legrain, M.; Dubus, P.; Karayan-Tapon, L.; Mosser, J.; Ichimura, K.; Figarella-Branger, D. Outcome-based determination of optimal pyrosequencing assay for mgmt methylation detection in glioblastoma patients. J. Neurooncol. 2014, 116, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.; Baborie, A.; Alam, F.; Joyce, K.; Moxham, M.; Sibson, R.; Crooks, D.; Husband, D.; Shenoy, A.; Brodbelt, A.; et al. Extent of mgmt promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br. J. Cancer 2009, 101, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, G.; Hentschel, B.; Felsberg, J.; Schackert, G.; Simon, M.; Schnell, O.; Westphal, M.; Wick, W.; Pietsch, T.; Loeffler, M.; et al. Predictive impact of mgmt promoter methylation in glioblastoma of the elderly. Int. J. Cancer. 2012, 131, 1342–1350. [Google Scholar]
- Oberstadt, M.C.; Bien-Moller, S.; Weitmann, K.; Herzog, S.; Hentschel, K.; Rimmbach, C.; Vogelgesang, S.; Balz, E.; Fink, M.; Michael, H.; et al. Epigenetic modulation of the drug resistance genes mgmt, abcb1 and abcg2 in glioblastoma multiforme. BMC Cancer 2013, 13, 617. [Google Scholar] [CrossRef] [PubMed]
- Everhard, S.; Tost, J.; el Abdalaoui, H.; Criniere, E.; Busato, F.; Marie, Y.; Gut, I.G.; Sanson, M.; Mokhtari, K.; Laigle-Donadey, F.; et al. Identification of regions correlating mgmt promoter methylation and gene expression in glioblastomas. Neuro-oncology 2009, 11, 348–356. [Google Scholar]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Delpu, Y.; Cordelier, P.; Cho, W.C.; Torrisani, J. DNA methylation and cancer diagnosis. Int. J. Mol. Sci. 2013, 14, 15029–15058. [Google Scholar] [CrossRef] [PubMed]
- Heichman, K.A.; Warren, J.D. DNA methylation biomarkers and their utility for solid cancer diagnostics. Clin. Chem. Lab. Med. 2012, 50, 1707–1721. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, K.; Liu, K. Epigenetics and colorectal cancer pathogenesis. Cancers 2013, 5, 676–713. [Google Scholar] [CrossRef] [PubMed]
- Colussi, D.; Brandi, G.; Bazzoli, F.; Ricciardiello, L. Molecular pathways involved in colorectal cancer: Implications for disease behavior and prevention. Int. J. Mol. Sci. 2013, 14, 16365–16385. [Google Scholar] [PubMed]
- Gyparaki, M.T.; Basdra, E.K.; Papavassiliou, A.G. DNA methylation biomarkers as diagnostic and prognostic tools in colorectal cancer. J. Mol. Med. (Berlin, Germany) 2013, 91, 1249–1256. [Google Scholar]
- Balgkouranidou, I.; Liloglou, T.; Lianidou, E.S. Lung cancer epigenetics: Emerging biomarkers. Biomark. Med. 2013, 7, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.; Lechner, M.; Fourkala, E.O.; Kristeleit, R.; Widschwendter, M. Emerging promise of epigenetics and DNA methylation for the diagnosis and management of women’s cancers. Epigenomics 2010, 2, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Day, T.K.; Bianco-Miotto, T. Common gene pathways and families altered by DNA methylation in breast and prostate cancers. Endocr. Relat. Cancer 2013, 20, R215–R232. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; van Tilborg, A.A.; Zwarthoff, E.C. DNA methylation-based biomarkers in bladder cancer. Nat. Rev. Urol. 2013, 10, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Fukushige, S.; Horii, A. Road to early detection of pancreatic cancer: Attempts to utilize epigenetic biomarkers. Cancer Lett. 2014, 342, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, E.S.; Chong, K.K.; Huynh, K.T.; Tanaka, R.; Hoon, D.S. Epigenetic biomarkers in skin cancer. Cancer Lett. 2014, 342, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Zoratto, F.; Rossi, L.; Verrico, M.; Papa, A.; Basso, E.; Zullo, A.; Tomao, L.; Romiti, A.; Lo Russo, G.; Tomao, S. Focus on genetic and epigenetic events of colorectal cancer pathogenesis: Implications for molecular diagnosis. Tumour Biol. 2014, 35, 6195–6206. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, M.A. Clinical applications of epigenetic markers and epigenetic profiling in myeloid malignancies. Semin. Oncol. 2012, 39, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vicente, A.E.; Diaz, M.G.; Hernandez-Rivas, J.M. Chronic lymphocytic leukemia: A clinical and molecular heterogenous disease. Cancer Genet. 2013, 206, 49–62. [Google Scholar]
- Li, Y.S.; Xie, Q.; Yang, D.Y.; Zheng, Y. Role of rassf1a promoter methylation in the pathogenesis of hepatocellular carcinoma: A meta-analysis of 21 cohort studies. Mol. Biol. Rep. 2014, 41, 3925–3933. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.D.; Han, Z.J.; Skoletsky, J.; Olson, J.; Sah, J.; Myeroff, L.; Platzer, P.; Lu, S.; Dawson, D.; Willis, J.; et al. Detection in fecal DNA of colon cancer-specific methylation of the nonexpressed vimentin gene. J. Natl. Cancer Inst. 2005, 97, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- deVos, T.; Tetzner, R.; Model, F.; Weiss, G.; Schuster, M.; Distler, J.; Steiger, K.V.; Grutzmann, R.; Pilarsky, C.; Habermann, J.K.; et al. Circulating methylated sept9 DNA in plasma is a biomarker for colorectal cancer. Clin. Chem. 2009, 55, 1337–1346. [Google Scholar]
- Oh, T.; Kim, N.; Moon, Y.; Kim, M.S.; Hoehn, B.D.; Park, C.H.; Kim, T.S.; Kim, N.K.; Chung, H.C.; An, S. Genome-wide identification and validation of a novel methylation biomarker, sdc2, for blood-based detection of colorectal cancer. J. Mol. Diagn. 2013, 15, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.M.; Ross, J.P.; Drew, H.R.; Ho, T.; Brown, G.S.; Saunders, N.F.; Duesing, K.R.; Buckley, M.J.; Dunne, R.; Beetson, I.; et al. A panel of genes methylated with high frequency in colorectal cancer. BMC Cancer 2014, 14, 54. [Google Scholar] [CrossRef] [PubMed]
- Goessl, C.; Krause, H.; Muller, M.; Heicappell, R.; Schrader, M.; Sachsinger, J.; Miller, K. Fluorescent methylation-specific polymerase chain reaction for DNA-based detection of prostate cancer in bodily fluids. Cancer Res. 2000, 60, 5941–5945. [Google Scholar] [PubMed]
- Goessl, C.; Muller, M.; Heicappell, R.; Krause, H.; Straub, B.; Schrader, M.; Miller, K. DNA-based detection of prostate cancer in urine after prostatic massage. Urology 2001, 58, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Merlo, A.; Herman, J.G.; Mao, L.; Lee, D.J.; Gabrielson, E.; Burger, P.C.; Baylin, S.B.; Sidransky, D. 5’ cpg island methylation is associated with transcriptional silencing of the tumour suppressor p16/cdkn2/mts1 in human cancers. Nat. Med. 1995, 1, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Sterlacci, W.; Tzankov, A.; Veits, L.; Zelger, B.; Bihl, M.P.; Foerster, A.; Augustin, F.; Fiegl, M.; Savic, S. A comprehensive analysis of p16 expression, gene status, and promoter hypermethylation in surgically resected non-small cell lung carcinomas. J. Thorac. Oncol. 2011, 6, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Nimmrich, I.; Sieuwerts, A.M.; Meijer-van Gelder, M.E.; Schwope, I.; Bolt-de Vries, J.; Harbeck, N.; Koenig, T.; Hartmann, O.; Kluth, A.; Dietrich, D.; et al. DNA hypermethylation of pitx2 is a marker of poor prognosis in untreated lymph node-negative hormone receptor-positive breast cancer patients. Breast Cancer Res. Treat. 2008, 111, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Nimmrich, I.; Hartmann, A.; Ross, J.S.; Cufer, T.; Grutzmann, R.; Kristiansen, G.; Paradiso, A.; Hartmann, O.; Margossian, A.; et al. Multicenter study using paraffin-embedded tumor tissue testing pitx2 DNA methylation as a marker for outcome prediction in tamoxifen-treated, node-negative breast cancer patients. J. Clin. Oncol. 2008, 26, 5036–5042. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.; Nimmrich, I.; Koenig, T.; Eppenberger-Castori, S.; Bohlmann, I.; Paradiso, A.; Spyratos, F.; Thomssen, C.; Mueller, V.; Nahrig, J.; et al. DNA-methylation of the homeodomain transcription factor PITX2 reliably predicts risk of distant disease recurrence in tamoxifen-treated, node-negative breast cancer patients—Technical and clinical validation in a multi-centre setting in collaboration with the European Organisation for Research and Treatment of Cancer (EORTC) PathoBiology group. Eur. J. Cancer 2007, 43, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, O.; Spyratos, F.; Harbeck, N.; Dietrich, D.; Fassbender, A.; Schmitt, M.; Eppenberger-Castori, S.; Vuaroqueaux, V.; Lerebours, F.; Welzel, K.; et al. DNA methylation markers predict outcome in node-positive, estrogen receptor-positive breast cancer with adjuvant anthracycline-based chemotherapy. Clin. Cancer Res. 2009, 15, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Banez, L.L.; Sun, L.; van Leenders, G.J.; Wheeler, T.M.; Bangma, C.H.; Freedland, S.J.; Ittmann, M.M.; Lark, A.L.; Madden, J.F.; Hartman, A.; et al. Multicenter clinical validation of PITX2 methylation as a prostate specific antigen recurrence predictor in patients with post-radical prostatectomy prostate cancer. J. Urol. 2010, 184, 149–156. [Google Scholar]
- Dietrich, D.; Hasinger, O.; Banez, L.L.; Sun, L.; van Leenders, G.J.; Wheeler, T.M.; Bangma, C.H.; Wernert, N.; Perner, S.; Freedland, S.J.; et al. Development and clinical validation of a real-time PCR assay for PITX2 DNA methylation to predict prostate-specific antigen recurrence in prostate cancer patients following radical prostatectomy. J. Mol. Diagn. 2013, 15, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Cottrell, S.; Distler, J.; Schatz, P.; Kristiansen, G.; Ittmann, M.; Haefliger, C.; Lesche, R.; Hartmann, A.; Corman, J.; et al. DNA methylation of the PITX2 gene promoter region is a strong independent prognostic marker of biochemical recurrence in patients with prostate cancer after radical prostatectomy. J. Urol. 2009, 181, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Salehi, F.; Scheithauer, B.W.; Rotondo, F.; Syro, L.V.; Kovacs, K. Role of MGMT in tumor development, progression, diagnosis, treatment and prognosis. Anticancer Res. 2009, 29, 3759–3768. [Google Scholar] [PubMed]
- Esteller, M.; Gaidano, G.; Goodman, S.N.; Zagonel, V.; Capello, D.; Botto, B.; Rossi, D.; Gloghini, A.; Vitolo, U.; Carbone, A.; et al. Hypermethylation of the DNA repair gene O(6)-methylguanine DNA methyltransferase and survival of patients with diffuse large B-cell lymphoma. J. Natl. Cancer Inst. 2002, 94, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Van den Bent, M.J.; Dubbink, H.J.; Sanson, M.; van der Lee-Haarloo, C.R.; Hegi, M.; Jeuken, J.W.; Ibdaih, A.; Brandes, A.A.; Taphoorn, M.J.; Frenay, M.; et al. MGMT promoter methylation is prognostic but not predictive for outcome to adjuvant PCV chemotherapy in anaplastic oligodendroglial tumors: A report from EORTC Brain Tumor Group Study 26951. J. Clin. Oncol. 2009, 27, 5881–5886. [Google Scholar]
- Wick, W.; Hartmann, C.; Engel, C.; Stoffels, M.; Felsberg, J.; Stockhammer, F.; Sabel, M.C.; Koeppen, S.; Ketter, R.; Meyermann, R.; et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J. Clin. Oncol. 2009, 27, 5874–5880. [Google Scholar] [PubMed]
- Skaar, D.A.; Li, Y.; Bernal, A.J.; Hoyo, C.; Murphy, S.K.; Jirtle, R.L. The human imprintome: Regulatory mechanisms, methods of ascertainment, and roles in disease susceptibility. ILAR J. 2012, 53, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 2014, 15, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G. Genomic imprinting disorders in humans: A mini-review. J. Assist. Reprod. Genet. 2009, 26, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, S.C.; Clayton-Smith, J.; Birch, R.; Buiting, K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med. Genet. 2010, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.; Hawkins, M.; Barton, D.E.; Meaney, K.; Guitart, M.; O’Grady, A.; Tobi, S.; Ramsden, S.C.; Elles, R.; Gray, E.; et al. Establishment of the first WHO international genetic reference panel for Prader Willi and Angelman syndromes. Eur. J. Hum. Genet. 2011, 19, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Das, S.; Christian, S.L.; Baylin, S.B.; Herman, J.G.; Ledbetter, D.H. Methylation-specific PCR simplifies imprinting analysis. Nat. Genet. 1997, 16, 16–17. [Google Scholar] [PubMed]
- Zeschnigk, M.; Lich, C.; Buiting, K.; Doerfler, W.; Horsthemke, B. A single-tube PCR test for the diagnosis of Angelman and Prader-Willi syndrome based on allelic methylation differences at the SNRPN locus. Eur. J. Hum. Genet. 1997, 5, 94–98. [Google Scholar] [PubMed]
- Henkhaus, R.S.; Kim, S.J.; Kimonis, V.E.; Gold, J.A.; Dykens, E.M.; Driscoll, D.J.; Butler, M.G. Methylation-specific multiplex ligation-dependent probe amplification and identification of deletion genetic subtypes in Prader-Willi syndrome. Genet. Test. Mol. Biomark. 2012, 16, 178–186. [Google Scholar] [CrossRef]
- Bourque, D.K.; Penaherrera, M.S.; Yuen, R.K.; van Allen, M.I.; McFadden, D.E.; Robinson, W.P. The utility of quantitative methylation assays at imprinted genes for the diagnosis of fetal and placental disorders. Clin. Genet. 2011, 79, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Eggermann, T.; Begemann, M.; Binder, G.; Spengler, S. Silver-Russell syndrome: Genetic basis and molecular genetic testing. Orphanet J. Rare Dis. 2010, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Weksberg, R.; Shuman, C.; Beckwith, J.B. Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2010, 18, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Smilinich, N.J.; Day, C.D.; Fitzpatrick, G.V.; Caldwell, G.M.; Lossie, A.C.; Cooper, P.R.; Smallwood, A.C.; Joyce, J.A.; Schofield, P.N.; Reik, W.; et al. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 8064–8069. [Google Scholar] [CrossRef] [PubMed]
- Horike, S.; Ferreira, J.C.; Meguro-Horike, M.; Choufani, S.; Smith, A.C.; Shuman, C.; Meschino, W.; Chitayat, D.; Zackai, E.; Scherer, S.W.; et al. Screening of DNA methylation at the H19 promoter or the distal region of its ICR1 ensures efficient detection of chromosome 11p15 epimutations in Russell-Silver syndrome. Am. J. Med. Genet. A 2009, 149A, 2415–2423. [Google Scholar]
- Lukova, M.; Todorova, A.; Todorov, T.; Mitev, V. Different methylation patterns in BWS/SRS cases clarified by MS-MLPA. Mol. Biol. Rep. 2013, 40, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Priolo, M.; Sparago, A.; Mammi, C.; Cerrato, F.; Lagana, C.; Riccio, A. MS-MLPA is a specific and sensitive technique for detecting all chromosome 11p15.5 imprinting defects of BWS and SRS in a single-tube experiment. Eur. J. Hum. Genet. 2008, 16, 565–571. [Google Scholar]
- Scott, R.H.; Douglas, J.; Baskcomb, L.; Nygren, A.O.; Birch, J.M.; Cole, T.R.; Cormier-Daire, V.; Eastwood, D.M.; Garcia-Minaur, S.; Lupunzina, P.; et al. Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) robustly detects and distinguishes 11p15 abnormalities associated with overgrowth and growth retardation. J. Med. Genet. 2008, 45, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Calvello, M.; Tabano, S.; Colapietro, P.; Maitz, S.; Pansa, A.; Augello, C.; Lalatta, F.; Gentilin, B.; Spreafico, F.; Calzari, L.; et al. Quantitative DNA methylation analysis improves epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. Epigenetics 2013, 8, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- White, H.E.; Durston, V.J.; Harvey, J.F.; Cross, N.C. Quantitative analysis of SNRPN(correction of SRNPN) gene methylation by pyrosequencing as a diagnostic test for Prader-Willi syndrome and Angelman syndrome. Clin. Chem. 2006, 52, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2011, 13, 97–109. [Google Scholar]
- Mazzio, E.A.; Soliman, K.F. Basic concepts of epigenetics: Impact of environmental signals on gene expression. Epigenetics 2012, 7, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Cortessis, V.K.; Thomas, D.C.; Levine, A.J.; Breton, C.V.; Mack, T.M.; Siegmund, K.D.; Haile, R.W.; Laird, P.W. Environmental epigenetics: Prospects for studying epigenetic mediation of exposure-response relationships. Hum. Genet. 2012, 131, 1565–1589. [Google Scholar] [CrossRef] [PubMed]
- Hogg, K.; Price, E.M.; Hanna, C.W.; Robinson, W.P. Prenatal and perinatal environmental influences on the human fetal and placental epigenome. Clin. Pharmacol. Ther. 2012, 92, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Mirbahai, L.; Chipman, J.K. Epigenetic memory of environmental organisms: A reflection of lifetime stressor exposures. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 764–765, 10–17. [Google Scholar]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Dogan, M.V.; Shields, B.; Cutrona, C.; Gao, L.; Gibbons, F.X.; Simons, R.; Monick, M.; Brody, G.H.; Tan, K.; Beach, S.R.; et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics 2014, 15, 151. [Google Scholar] [CrossRef] [PubMed]
- Elliott, H.R.; Tillin, T.; McArdle, W.L.; Ho, K.; Duggirala, A.; Frayling, T.M.; Davey Smith, G.; Hughes, A.D.; Chaturvedi, N.; Relton, C.L. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin. Epigenetics 2014, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.R.; Haberg, S.E.; Nilsen, R.M.; Wang, X.; Vollset, S.E.; Murphy, S.K.; Huang, Z.; Hoyo, C.; Midttun, O.; Cupul-Uicab, L.A.; et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2012, 120, 1425–1431. [Google Scholar]
- Philibert, R.A.; Beach, S.R.; Lei, M.K.; Brody, G.H. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin. Epigenetics 2013, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Shenker, N.S.; Polidoro, S.; van Veldhoven, K.; Sacerdote, C.; Ricceri, F.; Birrell, M.A.; Belvisi, M.G.; Brown, R.; Vineis, P.; Flanagan, J.M. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum. Mol. Genet. 2013, 22, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Zeilinger, S.; Kuhnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One 2013, 8, e63812. [Google Scholar] [CrossRef] [PubMed]
- Monick, M.M.; Beach, S.R.; Plume, J.; Sears, R.; Gerrard, M.; Brody, G.H.; Philibert, R.A. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Besingi, W.; Johansson, A. Smoke-related DNA methylation changes in the etiology of human disease. Hum. Mol. Genet. 2014, 23, 2290–2297. [Google Scholar] [CrossRef] [PubMed]
- Markunas, C.A.; Xu, Z.; Harlid, S.; Wade, P.A.; Lie, R.T.; Taylor, J.A.; Wilcox, A.J. Identification of DNA Methylation Changes in Newborns Related to Maternal Smoking during Pregnancy. Environ. Health Perspect. 2014. [Google Scholar] [CrossRef]
- Harlid, S.; Xu, Z.; Panduri, V.; Sandler, D.P.; Taylor, J.A. CpG Sites Associated with Cigarette Smoking: Analysis of Epigenome-Wide Data from the Sister Study. Environ. Health Perspect. 2014, 122, 673–678. [Google Scholar] [PubMed]
- Novakovic, B.; Ryan, J.; Pereira, N.; Boughton, B.; Craig, J.M.; Saffery, R. Postnatal stability, tissue, and time specific effects of AHRR methylation change in response to maternal smoking in pregnancy. Epigenetics 2014, 9, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.R.; Haberg, S.E.; Bell, D.A.; Nilsen, R.M.; Vollset, S.E.; Midttun, O.; Ueland, P.M.; Wu, M.C.; Nystad, W.; Peddada, S.D.; et al. Maternal Smoking and DNA Methylation in Newborns: In Utero Effect or Epigenetic Inheritance? Cancer Epidemiol. Biomark. Prev. 2014. [Google Scholar] [CrossRef]
- Brokken, L.J.; Lundberg-Giwercman, Y.; Meyts, E.R.; Eberhard, J.; Stahl, O.; Cohn-Cedermark, G.; Daugaard, G.; Arver, S.; Giwercman, A. Association between polymorphisms in the aryl hydrocarbon receptor repressor gene and disseminated testicular germ cell cancer. Front. Endocrinol. 2013, 4, 4. [Google Scholar] [CrossRef]
- Cauchi, S.; Stucker, I.; Cenee, S.; Kremers, P.; Beaune, P.; Massaad-Massade, L. Structure and polymorphisms of human aryl hydrocarbon receptor repressor (AhRR) gene in a French population: Relationship with CYP1A1 inducibility and lung cancer. Pharmacogenetics 2003, 13, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Li, W.W.; Yang, B.W.; Tao, Z.H.; Sun, H.C.; Wang, L.; Xia, J.L.; Qin, L.X.; Tang, Z.Y.; Fan, J.; et al. Aryl hydrocarbon receptor nuclear translocator is associated with tumor growth and progression of hepatocellular carcinoma. Int. J. Cancer 2012, 130, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M.; Katoh, T.; Motoyama, H.; Sasaki, H.; Tsugane, S.; Ikenoue, T. Analysis of the AhR, ARNT, and AhRR gene polymorphisms: Genetic contribution to endometriosis susceptibility and severity. Fertil. Steril. 2005, 84, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Lupien, S.J.; McEwen, B.S.; Gunnar, M.R.; Heim, C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 2009, 10, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Meaney, M.J.; Szyf, M. Environmental programming of stress responses through DNA methylation: Life at the interface between a dynamic environment and a fixed genome. Dialogues Clin. Neurosci. 2005, 7, 103–123. [Google Scholar] [PubMed]
- Weaver, I.C.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Oberlander, T.F.; Weinberg, J.; Papsdorf, M.; Grunau, R.; Misri, S.; Devlin, A.M. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 2008, 3, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Hompes, T.; Izzi, B.; Gellens, E.; Morreels, M.; Fieuws, S.; Pexsters, A.; Schops, G.; Dom, M.; van Bree, R.; Freson, K.; et al. Investigating the influence of maternal cortisol and emotional state during pregnancy on the DNA methylation status of the glucocorticoid receptor gene (NR3C1) promoter region in cord blood. J. Psychiatr. Res. 2013, 47, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Conradt, E.; Lester, B.M.; Appleton, A.A.; Armstrong, D.A.; Marsit, C.J. The roles of DNA methylation of NR3C1 and 11beta-HSD2 and exposure to maternal mood disorder in utero on newborn neurobehavior. Epigenetics 2013, 8, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.M.; Ruf, M.; Gunter, H.M.; Dohrmann, K.; Schauer, M.; Meyer, A.; Elbert, T. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Transl. Psychiatry 2011, 1, e21. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, C.J.; D’Errico, N.C.; Stees, J.; Hughes, D.A. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics 2012, 7, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Daskalakis, N.P.; Lehrner, A.; Desarnaud, F.; Bader, H.N.; Makotkine, I.; Flory, J.D.; Bierer, L.M.; Meaney, M.J. Influences of Maternal and Paternal PTSD on Epigenetic Regulation of the Glucocorticoid Receptor Gene in Holocaust Survivor Offspring. Am. J. Psychiatry 2014, 171, 872–880. [Google Scholar] [PubMed]
- McGowan, P.O.; Sasaki, A.; D’Alessio, A.C.; Dymov, S.; Labonte, B.; Szyf, M.; Turecki, G.; Meaney, M.J. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 2009, 12, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Perroud, N.; Paoloni-Giacobino, A.; Prada, P.; Olie, E.; Salzmann, A.; Nicastro, R.; Guillaume, S.; Mouthon, D.; Stouder, C.; Dieben, K.; et al. Increased methylation of glucocorticoid receptor gene (NR3C1) in adults with a history of childhood maltreatment: A link with the severity and type of trauma. Transl. Psychiatry 2011, 1, e59. [Google Scholar] [CrossRef] [PubMed]
- Labonte, B.; Yerko, V.; Gross, J.; Mechawar, N.; Meaney, M.J.; Szyf, M.; Turecki, G. Differential glucocorticoid receptor exon 1(B), 1(C), and 1(H) expression and methylation in suicide completers with a history of childhood abuse. Biol. Psychiatry 2012, 72, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Tyrka, A.R.; Price, L.H.; Marsit, C.; Walters, O.C.; Carpenter, L.L. Childhood adversity and epigenetic modulation of the leukocyte glucocorticoid receptor: Preliminary findings in healthy adults. PLoS One 2012, 7, e30148. [Google Scholar] [CrossRef] [PubMed]
- Melas, P.A.; Wei, Y.; Wong, C.C.; Sjoholm, L.K.; Aberg, E.; Mill, J.; Schalling, M.; Forsell, Y.; Lavebratt, C. Genetic and epigenetic associations of MAOA and NR3C1 with depression and childhood adversities. Int. J. Neuropsychopharmacol. 2013, 16, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Flory, J.D.; Bierer, L.M.; Henn-Haase, C.; Lehrner, A.; Desarnaud, F.; Makotkine, I.; Daskalakis, N.P.; Marmar, C.R.; Meaney, M.J. Lower Methylation of Glucocorticoid Receptor Gene Promoter 1 in Peripheral Blood of Veterans with Posttraumatic Stress Disorder. Biol. Psychiatry 2014. [Google Scholar] [CrossRef]
- Labonte, B.; Azoulay, N.; Yerko, V.; Turecki, G.; Brunet, A. Epigenetic modulation of glucocorticoid receptors in posttraumatic stress disorder. Transl. Psychiatry 2014, 4, e368. [Google Scholar] [CrossRef] [PubMed]
- Bromer, C.; Marsit, C.J.; Armstrong, D.A.; Padbury, J.F.; Lester, B. Genetic and epigenetic variation of the glucocorticoid receptor (NR3C1) in placenta and infant neurobehavior. Dev. Psychobiol. 2013, 55, 673–683. [Google Scholar] [PubMed]
- Yehuda, R.; Daskalakis, N.P.; Desarnaud, F.; Makotkine, I.; Lehrner, A.L.; Koch, E.; Flory, J.D.; Buxbaum, J.D.; Meaney, M.J.; Bierer, L.M. Epigenetic Biomarkers as Predictors and Correlates of Symptom Improvement Following Psychotherapy in Combat Veterans with PTSD. Front. Psychiatry 2013, 4, 118. [Google Scholar] [CrossRef] [PubMed]
- Edelman, S.; Shalev, I.; Uzefovsky, F.; Israel, S.; Knafo, A.; Kremer, I.; Mankuta, D.; Kaitz, M.; Ebstein, R.P. Epigenetic and genetic factors predict women’s salivary cortisol following a threat to the social self. PLoS One 2012, 7, e48597. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.; Champagne, F.A.; Brown, S.E.; Dymov, S.; Sharma, S.; Meaney, M.J.; Szyf, M. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: Altering epigenetic marking later in life. J. Neurosci. 2005, 25, 11045–11054. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.; Meaney, M.J.; Szyf, M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc. Natl. Acad. Sci. USA 2006, 103, 3480–3485. [Google Scholar] [CrossRef] [PubMed]
- Essex, M.J.; Thomas Boyce, W.; Hertzman, C.; Lam, L.L.; Armstrong, J.M.; Neumann, S.M.; Kobor, M.S. Epigenetic Vestiges of Early Developmental Adversity: Childhood Stress Exposure and DNA Methylation in Adolescence. Child Dev. 2011. [Google Scholar] [CrossRef]
- Uddin, M.; Aiello, A.E.; Wildman, D.E.; Koenen, K.C.; Pawelec, G.; de Los Santos, R.; Goldmann, E.; Galea, S. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc. Natl. Acad. Sci. USA 2010, 107, 9470–9475. [Google Scholar] [CrossRef] [PubMed]
- Borghol, N.; Suderman, M.; McArdle, W.; Racine, A.; Hallett, M.; Pembrey, M.; Hertzman, C.; Power, C.; Szyf, M. Associations with early-life socio-economic position in adult DNA methylation. Int. J. Epidemiol. 2012, 41, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Klengel, T.; Conneely, K.N.; Smith, A.K.; Altmann, A.; Pace, T.W.; Rex-Haffner, M.; Loeschner, A.; Gonik, M.; Mercer, K.B.; et al. Childhood maltreatment is associated with distinct genomic and epigenetic profiles in posttraumatic stress disorder. Proc. Natl. Acad. Sci. USA 2013, 110, 8302–8307. [Google Scholar] [CrossRef] [PubMed]
- Naumova, O.Y.; Lee, M.; Koposov, R.; Szyf, M.; Dozier, M.; Grigorenko, E.L. Differential patterns of whole-genome DNA methylation in institutionalized children and children raised by their biological parents. Dev. Psychopathol. 2012, 24, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Harvey, N.C.; Sheppard, A.; Godfrey, K.M.; McLean, C.; Garratt, E.; Ntani, G.; Davies, L.; Murray, R.; Inskip, H.M.; Gluckman, P.D.; et al. Childhood Bone Mineral Content Is Associated With Methylation Status of the RXRA Promoter at Birth. J. Bone Miner. Res. 2014, 29, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Relton, C.L.; Groom, A.; St Pourcain, B.; Sayers, A.E.; Swan, D.C.; Embleton, N.D.; Pearce, M.S.; Ring, S.M.; Northstone, K.; Tobias, J.H.; et al. DNA methylation patterns in cord blood DNA and body size in childhood. PLoS One 2012, 7, e31821. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Beyan, H.; Down, T.A.; Hawa, M.I.; Maslau, S.; Aden, D.; Daunay, A.; Busato, F.; Mein, C.A.; Manfras, B.; et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet. 2011, 7, e1002300. [Google Scholar] [CrossRef] [PubMed]
- Clarke-Harris, R.; Wilkin, T.J.; Hosking, J.; Pinkney, J.; Jeffery, A.N.; Metcalf, B.S.; Godfrey, K.M.; Voss, L.D.; Lillycrop, K.A.; Burdge, G.C. Peroxisomal proliferator activated receptor-gamma-co-activator-1alpha promoter methylation in blood at 5–7 years predicts adiposity from 9 to 14 years (earlybird 50). Diabetes 2014, 63, 2528–2537. [Google Scholar]
- Nguyen, A.; Rauch, T.A.; Pfeifer, G.P.; Hu, V.W. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. 2010, 24, 3036–3051. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Meaburn, E.L.; Ronald, A.; Price, T.S.; Jeffries, A.R.; Schalkwyk, L.C.; Plomin, R.; Mill, J. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol. Psychiatry 2014, 19, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fang, Y.; Zhang, F.; Xu, M.; Zhang, J.; Yan, J.; Ju, W.; Brown, W.T.; Zhong, N. Hypermethylation of the enolase gene (ENO2) in autism. Eur. J. Pediatr. 2014, 173, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Berko, E.R.; Suzuki, M.; Beren, F.; Lemetre, C.; Alaimo, C.M.; Calder, R.B.; Ballaban-Gil, K.; Gounder, B.; Kampf, K.; Kirschen, J.; et al. Mosaic epigenetic dysregulation of ectodermal cells in autism spectrum disorder. PLoS Genet. 2014, 10, e1004402. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, M.R.; Rubin, R.A.; Falcone, T.; Ting, A.H.; Natowicz, M.R. Brain transcriptional and epigenetic associations with autism. PLoS One 2012, 7, e44736. [Google Scholar] [CrossRef] [PubMed]
- Ladd-Acosta, C.; Hansen, K.D.; Briem, E.; Fallin, M.D.; Kaufmann, W.E.; Feinberg, A.P. Common DNA methylation alterations in multiple brain regions in autism. Mol. Psychiatry 2013. [Google Scholar] [CrossRef]
- Sun, C.; Burgner, D.P.; Ponsonby, A.L.; Saffery, R.; Huang, R.C.; Vuillermin, P.J.; Cheung, M.; Craig, J.M. Effects of early-life environment and epigenetics on cardiovascular disease risk in children: Highlighting the role of twin studies. Pediatr. Res. 2013, 73, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.P.; Bruneau, B.G. Epigenetics and cardiovascular development. Annu. Rev. Physiol. 2012, 74, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Shirodkar, A.V.; Marsden, P.A. Epigenetics in cardiovascular disease. Curr. Opin. Cardiol. 2011, 26, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, B.; Irvin, M.R.; Sha, J.; Zhi, D.; Aslibekyan, S.; Absher, D.; Tiwari, H.K.; Kabagambe, E.K.; Ordovas, J.M.; Arnett, D.K. Epigenome-wide association study of fasting measures of glucose, insulin, and HOMA-IR in the Genetics of Lipid Lowering Drugs and Diet Network study. Diabetes 2014, 63, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Irvin, M.R.; Zhi, D.; Joehanes, R.; Mendelson, M.; Aslibekyan, S.; Claas, S.A.; Thibeault, K.S.; Patel, N.; Day, K.; Waite Jones, L.; et al. Epigenome-Wide Association Study of Fasting Blood Lipids in the Genetics of Lipid Lowering Drugs and Diet Network Study. Circulation 2014, 130, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Campion, J.; Milagro, F.I.; Martinez, J.A. Individuality and epigenetics in obesity. Obes. Rev. 2009, 10, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Toperoff, G.; Aran, D.; Kark, J.D.; Rosenberg, M.; Dubnikov, T.; Nissan, B.; Wainstein, J.; Friedlander, Y.; Levy-Lahad, E.; Glaser, B.; et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum. Mol. Genet. 2012, 21, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Cordero, P.; Campion, J.; Milagro, F.I.; Goyenechea, E.; Steemburgo, T.; Javierre, B.M.; Martinez, J.A. Leptin and TNF-alpha promoter methylation levels measured by MSP could predict the response to a low-calorie diet. J. Physiol. Biochem. 2011, 67, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Guintivano, J.; Arad, M.; Gould, T.D.; Payne, J.L.; Kaminsky, Z.A. Antenatal prediction of postpartum depression with blood DNA methylation biomarkers. Mol. Psychiatry 2014, 19, 633. [Google Scholar] [CrossRef]
- Wockner, L.F.; Noble, E.P.; Lawford, B.R.; Young, R.M.; Morris, C.P.; Whitehall, V.L.; Voisey, J. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl. Psychiatry 2014, 4, e339. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, M.; Bundo, M.; Koike, S.; Takizawa, R.; Kakiuchi, C.; Araki, T.; Kasai, K.; Iwamoto, K. Comprehensive DNA methylation analysis of peripheral blood cells derived from patients with first-episode schizophrenia. J. Hum. Genet. 2013, 58, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Camarillo, C.; Ping, Y.; Arana, T.B.; Zhao, H.; Thompson, P.M.; Xu, C.; Su, B.B.; Fan, H.; Ordonez, J.; et al. The DNA methylome and transcriptome of different brain regions in schizophrenia and bipolar disorder. PLoS One 2014, 9, e95875. [Google Scholar] [CrossRef] [PubMed]
- Aberg, K.A.; McClay, J.L.; Nerella, S.; Clark, S.; Kumar, G.; Chen, W.; Khachane, A.N.; Xie, L.; Hudson, A.; Gao, G.; et al. Methylome-wide association study of schizophrenia: Identifying blood biomarker signatures of environmental insults. JAMA Psychiatry 2014, 71, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleky, H.M.; Nohesara, S.; Ghadirivasfi, M.; Lambert, A.W.; Ahmadkhaniha, H.; Ozturk, S.; Wong, C.K.; Shafa, R.; Mostafavi, A.; Thiagalingam, S. DNA hypermethylation of serotonin transporter gene promoter in drug naive patients with schizophrenia. Schizophr. Res. 2014, 152, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, M.; Bundo, M.; Kasai, K.; Iwamoto, K. DNA methylation in schizophrenia: Progress and challenges of epigenetic studies. Genome Med. 2012, 4, 96. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Iwamoto, K. Comprehensive DNA methylation and hydroxymethylation analysis in the human brain and its implication in mental disorders. Neuropharmacology 2014, 80, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleky, H.M.; Smith, C.L.; Faraone, S.V.; Shafa, R.; Stone, W.; Glatt, S.J.; Tsuang, M.T. Methylomics in psychiatry: Modulation of gene-environment interactions may be through DNA methylation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2004, 127B, 51–59. [Google Scholar]
- Abdolmaleky, H.M.; Yaqubi, S.; Papageorgis, P.; Lambert, A.W.; Ozturk, S.; Sivaraman, V.; Thiagalingam, S. Epigenetic dysregulation of HTR2A in the brain of patients with schizophrenia and bipolar disorder. Schizophr. Res. 2011, 129, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Ghadirivasfi, M.; Nohesara, S.; Ahmadkhaniha, H.R.; Eskandari, M.R.; Mostafavi, S.; Thiagalingam, S.; Abdolmaleky, H.M. Hypomethylation of the serotonin receptor type-2A gene (HTR2A) at T102C polymorphic site in DNA derived from the saliva of patients with schizophrenia and bipolar disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 536–545. [Google Scholar] [CrossRef]
- Mill, J.; Tang, T.; Kaminsky, Z.; Khare, T.; Yazdanpanah, S.; Bouchard, L.; Jia, P.; Assadzadeh, A.; Flanagan, J.; Schumacher, A.; et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am. J. Hum. Genet. 2008, 82, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleky, H.M.; Cheng, K.H.; Russo, A.; Smith, C.L.; Faraone, S.V.; Wilcox, M.; Shafa, R.; Glatt, S.J.; Nguyen, G.; Ponte, J.F.; et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: A preliminary report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 134B, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Grayson, D.R.; Jia, X.; Chen, Y.; Sharma, R.P.; Mitchell, C.P.; Guidotti, A.; Costa, E. Reelin promoter hypermethylation in schizophrenia. Proc. Natl. Acad. Sci. USA 2005, 102, 9341–9346. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, Z.; Tochigi, M.; Jia, P.; Pal, M.; Mill, J.; Kwan, A.; Ioshikhes, I.; Vincent, J.B.; Kennedy, J.L.; Strauss, J.; et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol. Psychiatry 2012, 17, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, H.; Iwamoto, K.; Bundo, M.; Ueda, J.; Miyauchi, T.; Komori, A.; Kazuno, A.; Adati, N.; Kusumi, I.; Okazaki, Y.; et al. Hypermethylation of serotonin transporter gene in bipolar disorder detected by epigenome analysis of discordant monozygotic twins. Transl. Psychiatry 2011, 1, e24. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, M.N.; Pitt, J.; Craig, J.M. Going back to the future with Guthrie-powered epigenome-wide association studies. Genome Med. 2012, 4, 83. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.C.; Spector, T.D.; Bell, J.T. Using epigenome-wide association scans of DNA methylation in age-related complex human traits. Epigenomics 2012, 4, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jockel, K.H.; Erbel, R.; Muhleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Le, J.; Barnes, B.; Saedinia-Melnyk, S.; Zhou, L.; Shen, R.; Gunderson, K.L. Geneom-wide methylation profiling using Infinium assay. Epigenomics 2009, 1, 177–200. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, W.W. Biomarkers and ageing: The clock-watcher. Nature 2014, 508, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Stampfer, M. Lifestyle factors and microsatellite instability in colorectal cancer: The evolving field of molecular pathological epidemiology. J. Natl. Cancer Inst. 2010, 102, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Bishehsari, F.; Mahdavinia, M.; Vacca, M.; Malekzadeh, R.; Mariani-Costantini, R. Epidemiological transition of colorectal cancer in developing countries: Environmental factors, molecular pathways, and opportunities for prevention. World J. Gastroenterol. 2014, 20, 6055–6072. [Google Scholar] [CrossRef] [PubMed]
- Whelan, R.; Watts, R.; Orr, C.A.; Althoff, R.R.; Artiges, E.; Banaschewski, T.; Barker, G.J.; Bokde, A.L.; Buchel, C.; Carvalho, F.M.; et al. Neuropsychosocial profiles of current and future adolescent alcohol misusers. Nature 2014, 512, 185–189. [Google Scholar]
- Kim, D.S.; Lee, J.Y.; Lee, S.M.; Choi, J.E.; Cho, S.; Park, J.Y. Promoter methylation of the RGC32 gene in nonsmall cell lung cancer. Cancer 2011, 117, 590–596. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mikeska, T.; Craig, J.M. DNA Methylation Biomarkers: Cancer and Beyond. Genes 2014, 5, 821-864. https://doi.org/10.3390/genes5030821
Mikeska T, Craig JM. DNA Methylation Biomarkers: Cancer and Beyond. Genes. 2014; 5(3):821-864. https://doi.org/10.3390/genes5030821
Chicago/Turabian StyleMikeska, Thomas, and Jeffrey M. Craig. 2014. "DNA Methylation Biomarkers: Cancer and Beyond" Genes 5, no. 3: 821-864. https://doi.org/10.3390/genes5030821
APA StyleMikeska, T., & Craig, J. M. (2014). DNA Methylation Biomarkers: Cancer and Beyond. Genes, 5(3), 821-864. https://doi.org/10.3390/genes5030821