Architecture of Inherited Susceptibility to Colorectal Cancer: A Voyage of Discovery


Abstract
:1. Introduction
2. Early Models of Genetic Susceptibility
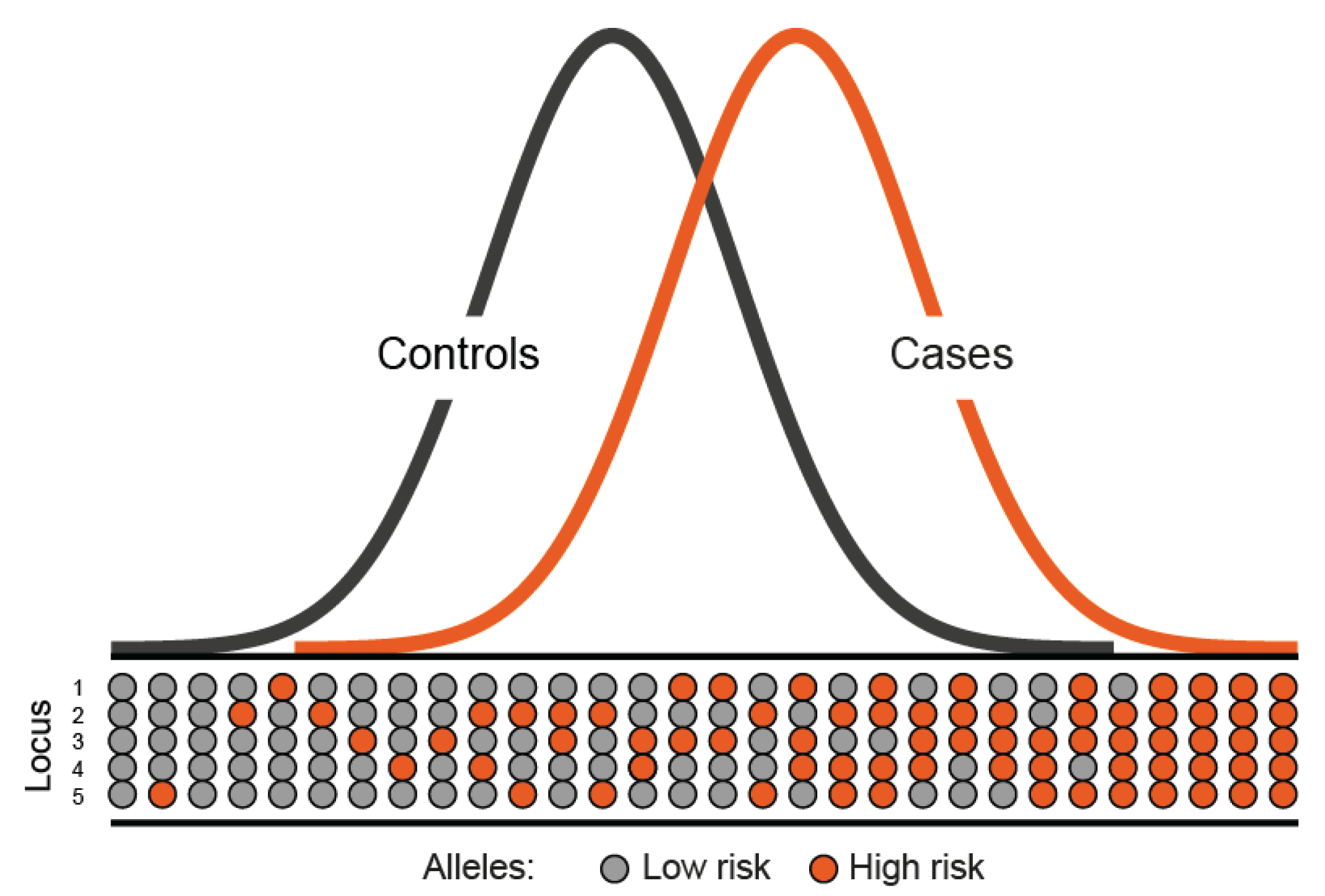
3. Identification of Rare High-Penetrance Susceptibility Alleles

{kind=link}
{kind=link}
| Gene(s) | Syndrome | Risk in mutation carriers | Mode of inheritance | References |
|---|---|---|---|---|
| APC | FAP | 90% by age 45 | Dominant | [10,11,12,13,14] |
| Mismatch repair (MLH1/MSH2/MSH6/PMS2) | HNPCC/Lynch syndrome | 40%–80% by age 75 | Dominant | [15,16,17,18,19,20,21,22,23,24,25,26,32,33] |
| SMAD4/BMPR1A | JPS | 17%–68% by age 60 | Dominant | [26,27] |
| STK11 | PJS | 39% by age 70 | Dominant | [28] |
| MUTYH | MYH-associated polyposis | 35%–53% | Recessive | [34,35] |
| POLD1/POLE | Oligopolyposis | Dominant | [36] |
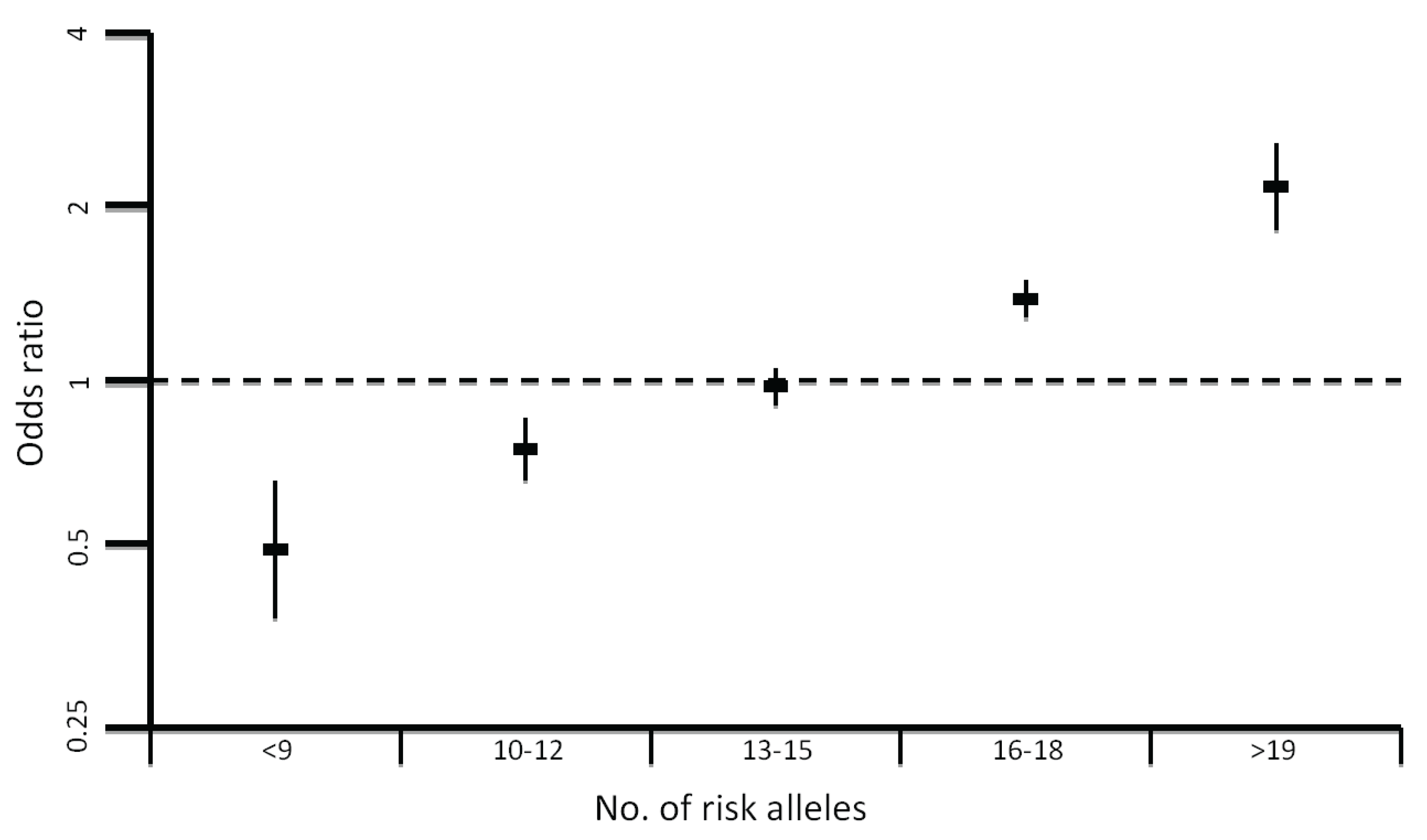
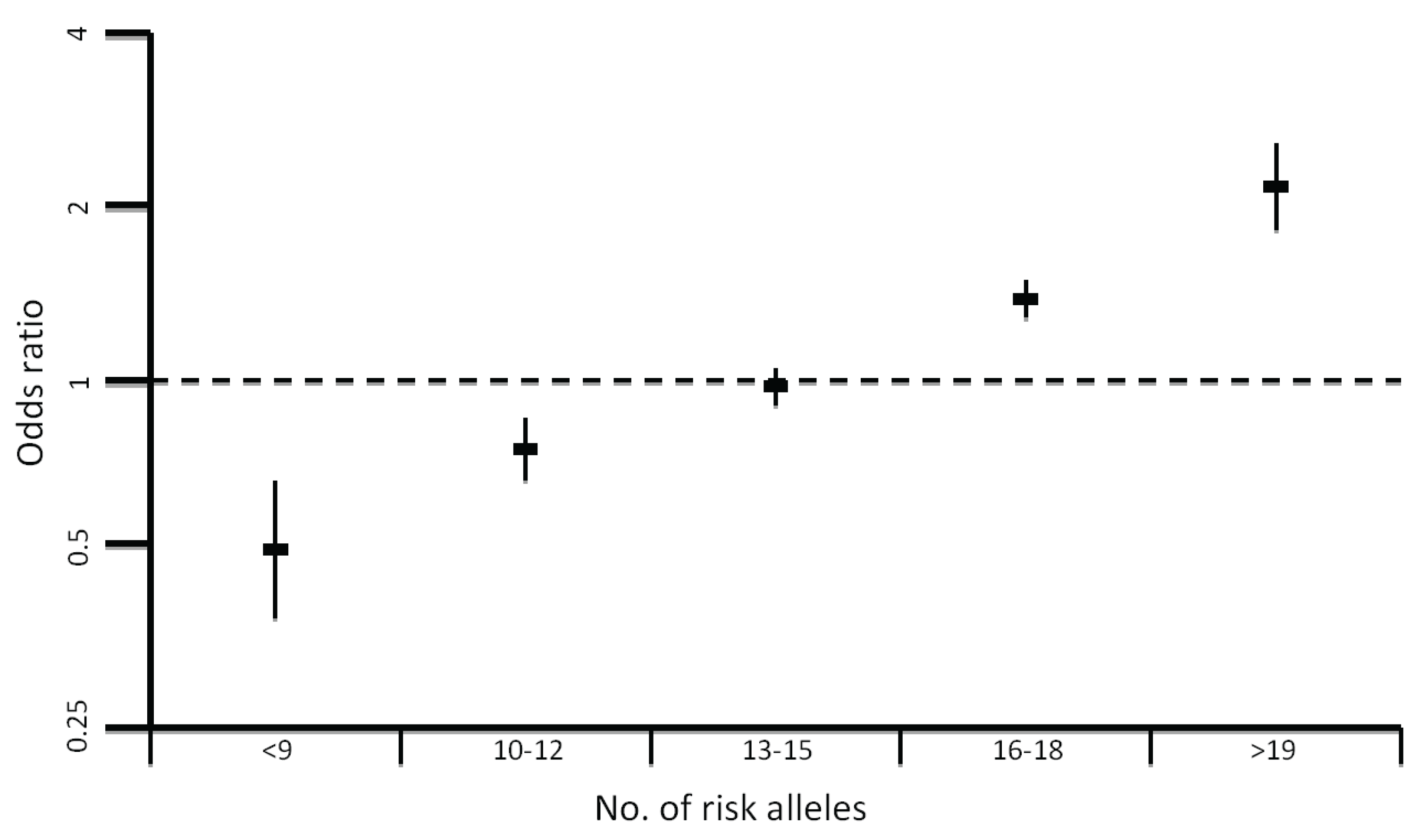
4. More Recent Models of Genetic Susceptibility
5. Rare, Moderately-Penetrant Disease-Causing Variants

6. Identification of Common Low-Penetrance Alleles
| Locus | Nearest Gene(s) | GWAS tagSNP | Location | Risk Allele | Alt Allele | RAF |
|---|---|---|---|---|---|---|
| 1q41 | DUSP10 | rs6691170 | 222,045,446 | T | G | 0.40 |
| 3q26.2 | TERC, MYNN | rs10936599 | 169,492,101 | C | T | 0.75 |
| 6p21.2 | CDKN1A | rs1321311 | 36,622,900 | T | G | 0.21 |
| 8q23.3 | EIF3H | rs16892766 | 117,630,683 | C | A | 0.09 |
| 8q24.21 | MYC | rs6983267 | 128,413,305 | G | T | 0.52 |
| 10p14 | GATA3 | rs10795668 | 8,701,219 | G | A | 0.67 |
| 11q13.4 | POLD3 | rs3824999 | 74,345,550 | C | A | 0.47 |
| 11q23.1 | FLJ45803 | rs3802842 | 111,171,709 | C | A | 0.27 |
| 12q13 | DIP2B, ATF1 | rs11169552 | 51,155,663 | C | T | 0.75 |
| 14q22.2 | BMP4 | rs4444235 | 54,410,919 | C | T | 0.48 |
| 15q13.3 | SCG5, GREM1 | rs4779584 | 32,994,756 | T | C | 0.19 |
| 16q22.1 | CDH1 | rs9929218 | 68,820,946 | G | A | 0.71 |
| 18q21.2 | SMAD7 | rs4939827 | 46,453,463 | T | C | 0.53 |
| 19q13.11 | RHPN2, GPATCH1 | rs10411210 | 33,532,300 | C | T | 0.90 |
| 20p12.3 | BMP2 | rs961253 | 6,404,281 | A | C | 0.37 |
| rs4813802 | 6,699,595 | G | T | 0.34 | ||
| 20q13.33 | LAMA5 | rs4925386 | 60,921,044 | C | T | 0.68 |
| Xp22.2 | SHROOM2 | rs5934683 | 9,751,474 | T | C | 0.56 |
7. Functional Effects of GWAS Loci
8. Impact of Common Variation on CRC Risk
9. Identifying Novel CRC Susceptibility Loci
10. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Smigal, C.; Thun, M.J. Cancer statistics, 2006. CA Cancer J. Clin. 2006, 56, 106–130. [Google Scholar] [CrossRef]
- Johns, L.E.; Houlston, R.S. A systematic review and meta-analysis of familial colorectal cancer risk. Am. J. Gastroenterol. 2001, 96, 2992–3003. [Google Scholar] [CrossRef]
- Woolf, C.M. A genetic study of carcinoma of the large intestine. Am. J. Hum. Genet. 1958, 10, 42–47. [Google Scholar]
- Macklin, M.T. Inheritance of cancer of the stomach and large intestine in man. J. Natl. Canc. Inst. 1960, 24, 551–571. [Google Scholar]
- Lovett, E. Familial factors in the etiology of carcinoma of the large bowel. Proc. R. Soc. Med. 1974, 67, 751–752. [Google Scholar]
- Ashley, D.J. Colonic cancer arising in polyposis coli. J. Med. Genet. 1969, 6, 376–378. [Google Scholar] [CrossRef]
- DeMars, R. 23rd Annual Symposium on Fundamental Cancer Research; Williams and Wilkins: Baltimore, MD, USA, 1969; pp. 105–106. [Google Scholar]
- Anderson, D.E. Genetic study of breast cancer: Identification of a high risk group. Cancer 1974, 34, 1090–1097. [Google Scholar] [CrossRef]
- Lynch, H.T.; Krush, A.J. Cancer family “G” Revisited: 1895–1970. Cancer 1971, 27, 1505–1511. [Google Scholar] [CrossRef]
- Bodmer, W.F.; Bailey, C.J.; Bodmer, J.; Bussey, H.J.; Ellis, A.; Gorman, P.; Lucibello, F.C.; Murday, V.A.; Rider, S.H.; Scambler, P.; et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 1987, 328, 614–616. [Google Scholar] [CrossRef]
- Leppert, M.; Dobbs, M.; Scambler, P.; O’Connell, P.; Nakamura, Y.; Stauffer, D.; Woodward, S.; Burt, R.; Hughes, J.; Gardner, E.; et al. The gene for familial polyposis coli maps to the long arm of chromosome 5. Science 1987, 238, 1411–1413. [Google Scholar]
- Herrera, L.; Kakati, S.; Gibas, L.; Pietrzak, E.; Sandberg, A.A. Gardner syndrome in a man with an interstitial deletion of 5q. Am. J. Med. Genet. 1986, 25, 473–476. [Google Scholar]
- Groden, J.; Thliveris, A.; Samowitz, W.; Carlson, M.; Gelbert, L.; Albertsen, H.; Joslyn, G.; Stevens, J.; Spirio, L.; Robertson, M.; et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991, 66, 589–600. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Nilbert, M.C.; Su, L.K.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hedge, P.; McKechnie, D.; et al. Identification of fap locus genes from chromosome 5q21. Science 1991, 253, 661–665. [Google Scholar]
- Miyaki, M.; Konishi, M.; Tanaka, K.; Kikuchi-Yanoshita, R.; Muraoka, M.; Yasuno, M.; Igari, T.; Koike, M.; Chiba, M.; Mori, T. Germline mutation of msh6 as the cause of hereditary nonpolyposis colorectal cancer. Nat. Genet. 1997, 17, 271–272. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Nicolaides, N.C.; Wei, Y.F.; Ruben, S.M.; Carter, K.C.; Rosen, C.A.; Haseltine, W.A.; Fleischmann, R.D.; Fraser, C.M.; Adams, M.D.; et al. Mutation of a mutl homolog in hereditary colon cancer. Science 1994, 263, 1625–1629. [Google Scholar]
- Bronner, C.E.; Baker, S.M.; Morrison, P.T.; Warren, G.; Smith, L.G.; Lescoe, M.K.; Kane, M.; Earabino, C.; Lipford, J.; Lindblom, A.; et al. Mutation in the DNA mismatch repair gene homologue hmlh1 is associated with hereditary non-polyposis colon cancer. Nature 1994, 368, 258–261. [Google Scholar] [CrossRef]
- Strand, M.; Prolla, T.A.; Liskay, R.M.; Petes, T.D. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 1993, 365, 274–276. [Google Scholar] [CrossRef]
- Peltomaki, P.; Aaltonen, L.A.; Sistonen, P.; Pylkkanen, L.; Mecklin, J.P.; Jarvinen, H.; Green, J.S.; Jass, J.R.; Weber, J.L.; Leach, F.S.; et al. Genetic mapping of a locus predisposing to human colorectal cancer. Science 1993, 260, 810–812. [Google Scholar]
- Aaltonen, L.A.; Peltomaki, P.; Leach, F.S.; Sistonen, P.; Pylkkanen, L.; Mecklin, J.P.; Jarvinen, H.; Powell, S.M.; Jen, J.; Hamilton, S.R.; et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260, 812–816. [Google Scholar]
- Lindblom, A.; Tannergard, P.; Werelius, B.; Nordenskjold, M. Genetic mapping of a second locus predisposing to hereditary non-polyposis colon cancer. Nat. Genet. 1993, 5, 279–282. [Google Scholar] [CrossRef]
- Leach, F.S.; Nicolaides, N.C.; Papadopoulos, N.; Liu, B.; Jen, J.; Parsons, R.; Peltomaki, P.; Sistonen, P.; Aaltonen, L.A.; Nystrom-Lahti, M.; et al. Mutations of a muts homolog in hereditary nonpolyposis colorectal cancer. Cell 1993, 75, 1215–1225. [Google Scholar] [CrossRef]
- Drummond, J.T.; Li, G.M.; Longley, M.J.; Modrich, P. Isolation of an hmsh2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science 1995, 268, 1909–1912. [Google Scholar]
- Palombo, F.; Gallinari, P.; Iaccarino, I.; Lettieri, T.; Hughes, M.; D’Arrigo, A.; Truong, O.; Hsuan, J.J.; Jiricny, J. Gtbp, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science 1995, 268, 1912–1914. [Google Scholar]
- Akiyama, Y.; Sato, H.; Yamada, T.; Nagasaki, H.; Tsuchiya, A.; Abe, R.; Yuasa, Y. Germ-line mutation of the hmsh6/gtbp gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Canc. Res. 1997, 57, 3920–3923. [Google Scholar]
- Shibata, D.; Peinado, M.A.; Ionov, Y.; Malkhosyan, S.; Perucho, M. Genomic instability in repeated sequences is an early somatic event in colorectal tumorigenesis that persists after transformation. Nat. Genet. 1994, 6, 273–281. [Google Scholar] [CrossRef]
- Howe, J.R.; Roth, S.; Ringold, J.C.; Summers, R.W.; Jarvinen, H.J.; Sistonen, P.; Tomlinson, I.P.; Houlston, R.S.; Bevan, S.; Mitros, F.A.; et al. Mutations in the smad4/dpc4 gene in juvenile polyposis. Science 1998, 280, 1086–1088. [Google Scholar] [CrossRef]
- Howe, J.R.; Bair, J.L.; Sayed, M.G.; Anderson, M.E.; Mitros, F.A.; Petersen, G.M.; Velculescu, V.E.; Traverso, G.; Vogelstein, B. Germline mutations of the gene encoding bone morphogenetic protein receptor 1a in juvenile polyposis. Nat. Genet. 2001, 28, 184–187. [Google Scholar] [CrossRef]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Hoglund, P.; et al. A serine/threonine kinase gene defective in peutz-jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef]
- Jarvinen, H.J.; Aarnio, M.; Mustonen, H.; Aktan-Collan, K.; Aaltonen, L.A.; Peltomaki, P.; de La Chapelle, A.; Mecklin, J.P. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 2000, 118, 829–834. [Google Scholar] [CrossRef]
- Burn, J.; Gerdes, A.M.; Macrae, F.; Mecklin, J.P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the capp2 randomised controlled trial. Lancet 2011, 378, 2081–2087. [Google Scholar] [CrossRef]
- Nicolaides, N.C.; Papadopoulos, N.; Liu, B.; Wei, Y.F.; Carter, K.C.; Ruben, S.M.; Rosen, C.A.; Haseltine, W.A.; Fleischmann, R.D.; Fraser, C.M.; et al. Mutations of two pms homologues in hereditary nonpolyposis colon cancer. Nature 1994, 371, 75–80. [Google Scholar] [CrossRef]
- Hendriks, Y.M.; Jagmohan-Changur, S.; van der Klift, H.M.; Morreau, H.; van Puijenbroek, M.; Tops, C.; van Os, T.; Wagner, A.; Ausems, M.G.; Gomez, E.; et al. Heterozygous mutations in pms2 cause hereditary nonpolyposis colorectal carcinoma (lynch syndrome). Gastroenterology 2006, 130, 312–322. [Google Scholar] [CrossRef]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R.; et al. Inherited variants of myh associated with somatic g:C-->t:A mutations in colorectal tumors. Nat. Genet. 2002, 30, 227–232. [Google Scholar]
- Lubbe, S.J.; di Bernardo, M.C.; Chandler, I.P.; Houlston, R.S. Clinical implications of the colorectal cancer risk associated with mutyh mutation. J. Clin. Oncol. 2009, 27, 3975–3980. [Google Scholar]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of pole and pold1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer—Analyses of cohorts of twins from sweden, denmark, and finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef]
- Lubbe, S.J.; Webb, E.L.; Chandler, I.P.; Houlston, R.S. Implications of familial colorectal cancer risk profiles and microsatellite instability status. J. Clin. Oncol. 2009, 27, 2238–2244. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Carvajal-Carmona, L.; Sellick, G.S.; Kemp, Z.; Webb, E.; Spain, S.; Sullivan, K.; Barclay, E.; Lubbe, S.; Jaeger, E.; et al. Deciphering the genetics of hereditary non-syndromic colorectal cancer. Eur. J. Hum. Genet. 2008, 16, 1477–1486. [Google Scholar] [CrossRef]
- Wiesner, G.L.; Daley, D.; Lewis, S.; Ticknor, C.; Platzer, P.; Lutterbaugh, J.; MacMillen, M.; Baliner, B.; Willis, J.; Elston, R.C.; et al. A subset of familial colorectal neoplasia kindreds linked to chromosome 9q22.2–31.2. Proc. Natl. Acad. Sci. USA 2003, 100, 12961–12965. [Google Scholar]
- Bodmer, W.; Bonilla, C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 2008, 40, 695–701. [Google Scholar]
- Laken, S.J.; Petersen, G.M.; Gruber, S.B.; Oddoux, C.; Ostrer, H.; Giardiello, F.M.; Hamilton, S.R.; Hampel, H.; Markowitz, A.; Klimstra, D.; et al. Familial colorectal cancer in ashkenazim due to a hypermutable tract in apc. Nat. Genet. 1997, 17, 79–83. [Google Scholar] [CrossRef]
- Whiffin, N.; Broderick, P.; Lubbe, S.J.; Pittman, A.M.; Penegar, S.; Chandler, I.; Houlston, R.S. Mlh1-93G>A is a risk factor for msi colorectal cancer. Carcinogenesis 2011, 32, 1157–1161. [Google Scholar] [CrossRef]
- Lipton, L.; Halford, S.E.; Johnson, V.; Novelli, M.R.; Jones, A.; Cummings, C.; Barclay, E.; Sieber, O.; Sadat, A.; Bisgaard, M.L.; et al. Carcinogenesis in myh-associated polyposis follows a distinct genetic pathway. Canc. Res. 2003, 63, 7595–7599. [Google Scholar]
- Reich, D.E.; Lander, E.S. On the allelic spectrum of human disease. Trends Genet. 2001, 17, 502–510. [Google Scholar] [CrossRef]
- Risch, N.; Merikangas, K. The future of genetic studies of complex human diseases. Science 1996, 273, 1516–1517. [Google Scholar]
- Broderick, P.; Carvajal-Carmona, L.; Pittman, A.M.; Webb, E.; Howarth, K.; Rowan, A.; Lubbe, S.; Spain, S.; Sullivan, K.; Fielding, S.; et al. A genome-wide association study shows that common alleles of smad7 influence colorectal cancer risk. Nat. Genet. 2007, 39, 1315–1317. [Google Scholar] [CrossRef]
- Dunlop, M.G.; Dobbins, S.E.; Farrington, S.M.; Jones, A.M.; Palles, C.; Whiffin, N.; Tenesa, A.; Spain, S.; Broderick, P.; Ooi, L.Y.; et al. Common variation near cdkn1a, pold3 and shroom2 influences colorectal cancer risk. Nat. Genet. 2012, 44, 770–776. [Google Scholar] [CrossRef]
- Houlston, R.S.; Cheadle, J.; Dobbins, S.E.; Tenesa, A.; Jones, A.M.; Howarth, K.; Spain, S.L.; Broderick, P.; Domingo, E.; Farrington, S.; et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat. Genet. 2010, 42, 973–977. [Google Scholar] [CrossRef]
- Houlston, R.S.; Webb, E.; Broderick, P.; Pittman, A.M.; di Bernardo, M.C.; Lubbe, S.; Chandler, I.; Vijayakrishnan, J.; Sullivan, K.; Penegar, S.; et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat. Genet. 2008, 40, 1426–1435. [Google Scholar] [CrossRef]
- Tenesa, A.; Farrington, S.M.; Prendergast, J.G.; Porteous, M.E.; Walker, M.; Haq, N.; Barnetson, R.A.; Theodoratou, E.; Cetnarskyj, R.; Cartwright, N.; et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat. Genet. 2008, 40, 631–637. [Google Scholar]
- Tomlinson, I.P.; Carvajal-Carmona, L.G.; Dobbins, S.E.; Tenesa, A.; Jones, A.M.; Howarth, K.; Palles, C.; Broderick, P.; Jaeger, E.E.; Farrington, S.; et al. Multiple common susceptibility variants near bmp pathway loci grem1, bmp4, and bmp2 explain part of the missing heritability of colorectal cancer. PLoS Genet. 2011, 7, e1002105. [Google Scholar] [CrossRef]
- Tomlinson, I.P.; Webb, E.; Carvajal-Carmona, L.; Broderick, P.; Howarth, K.; Pittman, A.M.; Spain, S.; Lubbe, S.; Walther, A.; Sullivan, K.; et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat. Genet. 2008, 40, 623–630. [Google Scholar] [CrossRef]
- Lubbe, S.J.; di Bernardo, M.C.; Broderick, P.; Chandler, I.; Houlston, R.S. Comprehensive evaluation of the impact of 14 genetic variants on colorectal cancer phenotype and risk. Am. J. Epidemiol. 2012, 175, 1–10. [Google Scholar] [CrossRef]
- Whiffin, N.; Dobbins, S.E.; Hosking, F.J.; Palles, C.; Tenesa, A.; Wang, Y.; Farrington, S.M.; Jones, A.M.; Broderick, P.; Campbell, H.; et al. Deciphering the genetic architecture of low-penetrance susceptibility to colorectal cancer. Hum. Mol. Genet. 2013, 22, 5075–5082. [Google Scholar] [CrossRef]
- Carvajal-Carmona, L.G.; Cazier, J.B.; Jones, A.M.; Howarth, K.; Broderick, P.; Pittman, A.; Dobbins, S.; Tenesa, A.; Farrington, S.; Prendergast, J.; et al. Fine-mapping of colorectal cancer susceptibility loci at 8q23.3, 16q22.1 and 19q13.11: Refinement of association signals and use of in silico analysis to suggest functional variation and unexpected candidate target genes. Hum. Mol. Genet. 2011, 20, 2879–2888. [Google Scholar] [CrossRef]
- Easton, D.F.; Pooley, K.A.; Dunning, A.M.; Pharoah, P.D.; Thompson, D.; Ballinger, D.G.; Struewing, J.P.; Morrison, J.; Field, H.; Luben, R.; et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007, 447, 1087–1093. [Google Scholar] [CrossRef]
- Pittman, A.M.; Naranjo, S.; Jalava, S.E.; Twiss, P.; Ma, Y.; Olver, B.; Lloyd, A.; Vijayakrishnan, J.; Qureshi, M.; Broderick, P.; et al. Allelic variation at the 8q23.3 colorectal cancer risk locus functions as a cis-acting regulator of eif3h. PLoS Genet. 2010, 6, e1001126. [Google Scholar]
- Tuupanen, S.; Turunen, M.; Lehtonen, R.; Hallikas, O.; Vanharanta, S.; Kivioja, T.; Bjorklund, M.; Wei, G.; Yan, J.; Niittymaki, I.; et al. The common colorectal cancer predisposition snp rs6983267 at chromosome 8q24 confers potential to enhanced wnt signaling. Nat. Genet. 2009, 41, 885–890. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Ahmadiyeh, N.; Jia, L.; Herman, P.; Verzi, M.P.; Doddapaneni, H.; Beckwith, C.A.; Chan, J.A.; Hills, A.; Davis, M.; et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with myc in colorectal cancer. Nat. Genet. 2009, 41, 882–884. [Google Scholar] [CrossRef]
- Pittman, A.M.; Webb, E.; Carvajal-Carmona, L.; Howarth, K.; di Bernardo, M.C.; Broderick, P.; Spain, S.; Walther, A.; Price, A.; Sullivan, K.; et al. Refinement of the basis and impact of common 11q23.1 variation to the risk of developing colorectal cancer. Hum. Mol. Genet. 2008, 17, 3720–3727. [Google Scholar] [CrossRef]
- Pittman, A.M.; Naranjo, S.; Webb, E.; Broderick, P.; Lips, E.H.; van Wezel, T.; Morreau, H.; Sullivan, K.; Fielding, S.; Twiss, P.; et al. The colorectal cancer risk at 18q21 is caused by a novel variant altering smad7 expression. Genome Res. 2009, 19, 987–993. [Google Scholar] [CrossRef]
- Ghoussaini, M.; Song, H.; Koessler, T.; Al Olama, A.A.; Kote-Jarai, Z.; Driver, K.E.; Pooley, K.A.; Ramus, S.J.; Kjaer, S.K.; Hogdall, E.; et al. Multiple loci with different cancer specificities within the 8q24 gene desert. J. Natl. Canc. Inst. 2008, 100, 962–966. [Google Scholar] [CrossRef]
- Kiemeney, L.A.; Thorlacius, S.; Sulem, P.; Geller, F.; Aben, K.K.; Stacey, S.N.; Gudmundsson, J.; Jakobsdottir, M.; Bergthorsson, J.T.; Sigurdsson, A.; et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat. Genet. 2008, 40, 1307–1312. [Google Scholar] [CrossRef]
- Crowther-Swanepoel, D.; Broderick, P.; di Bernardo, M.C.; Dobbins, S.E.; Torres, M.; Mansouri, M.; Ruiz-Ponte, C.; Enjuanes, A.; Rosenquist, R.; Carracedo, A.; et al. Common variants at 2q37.3, 8q24.21, 15q21.3 and 16q24.1 influence chronic lymphocytic leukemia risk. Nat. Genet. 2010, 42, 132–136. [Google Scholar] [CrossRef]
- Al Olama, A.A.; Kote-Jarai, Z.; Giles, G.G.; Guy, M.; Morrison, J.; Severi, G.; Leongamornlert, D.A.; Tymrakiewicz, M.; Jhavar, S.; Saunders, E.; et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat. Genet. 2009, 41, 1058–1060. [Google Scholar] [CrossRef]
- Amundadottir, L.T.; Sulem, P.; Gudmundsson, J.; Helgason, A.; Baker, A.; Agnarsson, B.A.; Sigurdsson, A.; Benediktsdottir, K.R.; Cazier, J.B.; Sainz, J.; et al. A common variant associated with prostate cancer in european and african populations. Nat. Genet. 2006, 38, 652–658. [Google Scholar] [CrossRef]
- Yeager, M.; Orr, N.; Hayes, R.B.; Jacobs, K.B.; Kraft, P.; Wacholder, S.; Minichiello, M.J.; Fearnhead, P.; Yu, K.; Chatterjee, N.; et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat. Genet. 2007, 39, 645–649. [Google Scholar] [CrossRef]
- Gudmundsson, J.; Sulem, P.; Manolescu, A.; Amundadottir, L.T.; Gudbjartsson, D.; Helgason, A.; Rafnar, T.; Bergthorsson, J.T.; Agnarsson, B.A.; Baker, A.; et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat. Genet. 2007, 39, 631–637. [Google Scholar] [CrossRef]
- Takatsuno, Y.; Mimori, K.; Yamamoto, K.; Sato, T.; Niida, A.; Inoue, H.; Imoto, S.; Kawano, S.; Yamaguchi, R.; Toh, H.; et al. The rs6983267 snp is associated with myc transcription efficiency, which promotes progression and worsens prognosis of colorectal cancer. Ann. Surg. Oncol. 2013, 20, 1395–1402. [Google Scholar] [CrossRef]
- Mrkonjic, M.; Roslin, N.M.; Greenwood, C.M.; Raptis, S.; Pollett, A.; Laird, P.W.; Pethe, V.V.; Chiang, T.; Daftary, D.; Dicks, E.; et al. Specific variants in the mlh1 gene region may drive DNA methylation, loss of protein expression, and msi-h colorectal cancer. PLoS One 2010, 5, e13314. [Google Scholar] [CrossRef]
- Wijnen, J.T.; Brohet, R.M.; van Eijk, R.; Jagmohan-Changur, S.; Middeldorp, A.; Tops, C.M.; van Puijenbroek, M.; Ausems, M.G.; Gomez Garcia, E.; Hes, F.J.; et al. Chromosome 8q23.3 and 11q23.1 variants modify colorectal cancer risk in lynch syndrome. Gastroenterology 2009, 136, 131–137. [Google Scholar] [CrossRef]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361, 361e1–361e2. [Google Scholar] [CrossRef]
- Eeles, R.A.; Olama, A.A.; Benlloch, S.; Saunders, E.J.; Leongamornlert, D.A.; Tymrakiewicz, M.; Ghoussaini, M.; Luccarini, C.; Dennis, J.; Jugurnauth-Little, S.; et al. Identification of 23 new prostate cancer susceptibility loci using the icogs custom genotyping array. Nat. Genet. 2013, 45, 385–391, 391e1–391e2. [Google Scholar] [CrossRef]
- Gylfe, A.E.; Katainen, R.; Kondelin, J.; Tanskanen, T.; Cajuso, T.; Hanninen, U.; Taipale, J.; Taipale, M.; Renkonen-Sinisalo, L.; Jarvinen, H.; et al. Eleven candidate susceptibility genes for common familial colorectal cancer. PLoS Genet. 2013, 9, e1003876. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.-P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capellá, G.; den Dunnen, J.T.; et al. Application of a 5-tiered scheme for standardized classification of 2360 unique mismatch repair gene variants in the insight locus-specific database. Nat. Genet. 2014, 46, 107–115. [Google Scholar]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the sift algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Lopez-Bigas, N. Improving the assessment of the outcome of nonsynonymous snvs with a consensus deleteriousness score, condel. Am. J. Hum. Genet. 2011, 88, 440–449. [Google Scholar] [CrossRef]
- Khurana, E.; Fu, Y.; Colonna, V.; Mu, X.J.; Kang, H.M.; Lappalainen, T.; Sboner, A.; Lochovsky, L.; Chen, J.; Harmanci, A.; et al. Integrative annotation of variants from 1092 humans: Application to cancer genomics. Science 2013, 342, 1235587. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Weedon, M.N.; Cebola, I.; Patch, A.M.; Flanagan, S.E.; de Franco, E.; Caswell, R.; Rodriguez-Segui, S.A.; Shaw-Smith, C.; Cho, C.H.; Lango Allen, H.; et al. Recessive mutations in a distal ptf1a enhancer cause isolated pancreatic agenesis. Nat. Genet. 2014, 46, 61–64. [Google Scholar]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. Tert promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Whiffin, N.; Houlston, R.S. Architecture of Inherited Susceptibility to Colorectal Cancer: A Voyage of Discovery. Genes 2014, 5, 270-284. https://doi.org/10.3390/genes5020270
Whiffin N, Houlston RS. Architecture of Inherited Susceptibility to Colorectal Cancer: A Voyage of Discovery. Genes. 2014; 5(2):270-284. https://doi.org/10.3390/genes5020270
Chicago/Turabian StyleWhiffin, Nicola, and Richard S. Houlston. 2014. "Architecture of Inherited Susceptibility to Colorectal Cancer: A Voyage of Discovery" Genes 5, no. 2: 270-284. https://doi.org/10.3390/genes5020270
APA StyleWhiffin, N., & Houlston, R. S. (2014). Architecture of Inherited Susceptibility to Colorectal Cancer: A Voyage of Discovery. Genes, 5(2), 270-284. https://doi.org/10.3390/genes5020270