Abstract
Cardiovascular diseases are a large contributor to causes of early death in developed countries. Some of these conditions, such as sudden cardiac death and atrial fibrillation, stem from arrhythmias—a spectrum of conditions with abnormal electrical activity in the heart. Genome-wide association studies can identify single nucleotide variations (SNVs) that may predispose individuals to developing acquired forms of arrhythmias. Through manual curation of published genome-wide association studies, we have collected a comprehensive list of 75 SNVs associated with cardiac arrhythmias. Ten of the SNVs result in amino acid changes and can be used in proteomic-based detection methods. In an effort to identify additional non-synonymous mutations that affect the proteome, we analyzed the post-translational modification S-nitrosylation, which is known to affect cardiac arrhythmias. We identified loss of seven known S-nitrosylation sites due to non-synonymous single nucleotide variations (nsSNVs). For predicted nitrosylation sites we found 1429 proteins where the sites are modified due to nsSNV. Analysis of the predicted S-nitrosylation dataset for over- or under-representation (compared to the complete human proteome) of pathways and functional elements shows significant statistical over-representation of the blood coagulation pathway. Gene Ontology (GO) analysis displays statistically over-represented terms related to muscle contraction, receptor activity, motor activity, cystoskeleton components, and microtubule activity. Through the genomic and proteomic context of SNVs and S-nitrosylation sites presented in this study, researchers can look for variation that can predispose individuals to cardiac arrhythmias. Such attempts to elucidate mechanisms of arrhythmia thereby add yet another useful parameter in predicting susceptibility for cardiac diseases.
1. Introduction
Cardiac arrhythmias encompass a range of conditions in which the normal rhythm of the heart is disrupted. The conditions include sudden cardiac death (SCD) and atrial fibrillation (AF), the most common form of arrhythmia [1,2]. The causes of arrhythmias such as AF are multifaceted and this presents difficulties in their treatment [3]. The number of Americans afflicted with arrhythmia is expected to increase as the population ages. Consequently, understanding the exact mechanisms of the disease and developing treatments is important.
DNA sequencing has evolved from its beginnings in the Sanger method. Now, next-generation sequencing (NGS) technologies allow the sequencing of millions of fragments of DNA in unison from a single sample. This process of parallel sequencing allows the whole genome to be sequenced in less than a day and is expected to be used in clinics in the near future [4]. Advances in DNA sequencing and genome-wide association studies are impacting detection, management, and treatment of diseases [5,6,7,8]. Several studies have investigated mutations and their associations with cardiac diseases. For example, long QT-syndrome (LQTS) is a disease characterized by prolonged repolarization of the heartbeat [9]. Research has shown that LQTS stems from mutations in genes encoding cardiac ion channels [10]. Over 100 mutations have been identified in 5 separate cardiac ion channel genes [9,10]. Other cardiac channelopathies, or disorders of the heart channel, affect normal cardiac rhythm in individuals [11]. Heritable cardiac arrhythmias, acquired from genetic alterations of these ion channels involved in cardiac heart rhythm, predispose affected individuals to sudden death [11]. Research on sudden cardiac death (SCD), which mainly results from severe ventricular arrhythmias, shows that a majority of mutations are found in the coding regions of ion channel units and key regulatory proteins. These mutations often lead to ion channel dysfunctions or affect biophysical properties of ion channels involved in normal heart function [12]. Further, computer modeling and simulations of cardiac myocytes have enabled scientists to represent variations in gene expression and allows them to reconstruct the effects of mutations characterized by functional changes in proteins [13]. Similar to DNA sequencing, proteomics technologies are also rapidly improving [14]. It is now possible to use proteomic technologies in conjunction with genomic technologies to detect and/or validate nsSNVs [15,16]. Such validation is critical to distinguish between inherited variations, somatic DNA and RNA modifications [17]. Therefore, having a list of amino acid changes that are potentially related to a specific disease can be of immense value as priority targets for proteomic validation.
One method for studying arrhythmias is investigating the genetic roots of the condition. This report presents the SNVs discovered or confirmed to have associations with arrhythmia over the past twenty years through genome-wide association and replication studies. Table 1 shows ten amino acid changing mutations, which can be detected using modern proteomic technologies [18]. While the variations are contained within a range of genes, the report further investigates two genes in particular. The first is NOS1AP, the most represented gene in Table S1, with 12 SNPs. The second gene is KCNN3, which hosts one of the exonic variants.

Table 1.
Summary of variant information from Table S1.
| Gene | Number of Variations | Chromosome Number | Amino Acid Change | Gene Description |
|---|---|---|---|---|
| SCN10A | 2 | 3 | V1073A | Sodium ion channel |
| HEY2 | 1 | 6 | 0 | Cardiovascular helix-loop-helix factor 1 |
| SCN5A | 5 | 3 | S1103Y, H558R | Sodium ion channel |
| TBX5 | 1 | 12 | 0 | T-box transcription factor |
| NOS1AP | 12 | 1 | 0 | Nitric oxide synthase 1 adaptor protein |
| ATP1B1 | 2 | 1 | 0 | Sodium/potassium-transporting ATPase subunit beta-1 |
| ZFHX3 | 3 | 16 | 0 | Zinc finger homeobox protein 3 |
| KCNN3 | 3 | 1 | N44N | Small conductance potassium channel |
| KCNJ2 | 2 | 17 | 0 | Potassium ion channel |
| XYLB | 3 | 3 | 0 | Energy metabolism |
| EXOG | 3 | 3 | 0 | Endonuclease |
| ACVR2B | 3 | 3 | 0 | Activin Receptor |
| RNF207 | 1 | 1 | G603A | Ring finger protein |
| PLN | 2 | 6 | 0 | Cardiac Muscle |
| KCNH2 | 5 | 7 | K897T, K557T | Potassium voltage gated channel |
| KCNQ1 | 4 | 11 | G643S | Potassium voltage gated channel |
| LITAF | 1 | 16 | 0 | DNA binding protein |
| NDRG4 | 1 | 16 | 0 | Mitogenic signalling |
| AGTR1 | 1 | 3 | 0 | Angiotensis II receptor |
| KNG1 | 1 | 3 | 0 | Kininogen |
| KCNE1 | 3 | 21 | D85N, S38G | Potassium voltage gated channel |
| KCNE4 | 1 | 2 | D196E | Potassium voltage-gated channel |
In an attempt to identify more amino acid changing variations potentially connected to the disease, we investigated the post-translational modification (PTM) nitrosylation. S-nitrosylation involves covalent attachment of Nitric Oxide (NO) to the thiol side chain of cysteine to form an S-nitrosothiol (SNO) [19]. NO is a well-known signaling molecule in the cardiovascular system and its role in cardiovascular diseases has been established [20]. S-nitrosylation is a reversible and selective PTM which regulates protein activity, cellular signal transduction, localization, and stability [19,21]. Modified levels of SNO proteins in the blood have been associated with high risks in patients with cardiovascular diseases [19]. Several studies have investigated these associations. Massy et al. found elevated plasma SNO levels in patients undergoing chronic hemodialysis predict cardiovascular outcomes [22]. Another study investigated NO as an endocrine vasoregulator in red blood cells and the potential impact NO plays in congestive heart failure [23]. In addition to nitrosylation other PTMs, such as glycosylation and phosphorylation, are also known to maintain the cardiac rhythm [12]. Current studies examine the association between acquired arrhythmias and PTMs of the cardiac sodium channel, which is involved in cardiac action potentials [24]. Cutler et al. demonstrated that neuronal nitric oxide synthase (NOS1) inhibition in the intact heart, along with the presence of increased myocardial Ca2+, increases an individual’s chances of Ca2+-mediated triggered arrhythmias [25]. Based on the aforementioned connections of nitrosylation and heart disease, we analyzed experimental and predicted loss of S-nitrosylation sites that are affected by nsSNVs.
2. Experimental
Manual curation of scientific articles retrieved using PubMed database [26] produced the 75 SNPs shown in Table S1. The articles selected from the PubMed database were found using search terms that included “Arrhythmia”, “Genome-wide”, “Association”, “Study”, “Cardiac”, and “SNP”. The articles published in the last 20 years were examined for reports of SNVs associated with cardiac arrhythmias. In these articles, SNVs discovered to have statistically significant associations with cardiac arrhythmias were added to the data shown in Table S1. To ensure consistency, articles that reported SNVs as a result of meta-analysis studies were not included in the list. Only SNVs that were discovered or confirmed in studies by the publishing group were eligible for inclusion. Of those SNVs, only those with statistically significant associations with cardiac arrhythmias were included. After extraction, background information on each SNV was collated from the Single Nucleotide Polymorphism Database or dbSNP [26] (Table 1 and Table S1). This public domain archive hosts a broad collection of simple genetic variations.
The seven experimentally verified S-nitrosylated cysteine sites that are affected by nsSNVs (Table 2) were obtained through mapping of S-nitrosylated proteins to the variations. These S-nitrosylated proteins were obtained from a literature review of endogenously S-nitrosylated proteins by Gould et al. [27]. Table 2 displays the proteins affected by the nsSNVs. Prediction of S-nitrosylation sites was performed as follows: the complete human proteome was obtained from UniProtKB/Swiss-Prot and GPS-SNO tool was used to predict N-nitrosylation sites [28]; pairwise alignments between Homo sapiens proteins and Mus musculus, Drosophila melanogaster, Arabidopsis thaliana and Saccharomyces cerevisiae proteins was performed followed by mapping of nsSNVs from SNVDis [29] to create a table that included predicted S-nitrosylation sites, conserved sites among the species and variation. Enrichment analysis was performed to identify over- or under-represented pathways or GO terms in the protein dataset, compared to their occurrence in the complete human proteome (Table 3). The expected occurrence of a pathway or GO term was calculated based on the actual number of times it was present in the human proteome [29,30]. Over- and under-represented pathways and processes were compared through significance in p values calculated based on methods described earlier [31].
Table 2.
Loss of experimentally confirmed S-nitrosylation sites by nsSNVs.
| Protein name | Position | Variation | Subseq | Ortholog | Species |
|---|---|---|---|---|---|
| Alpha-enolase | 357 | c->y | qackl | P21550 | Mouse |
| Cysteine and glycine-rich protein 3 | 58 | c->g | iyckv | P50462 | Mouse |
| Myosin-6 | 949 | c->y | decse | Q02566 | Mouse |
| Catenin beta-1 | 619 | c->y | vlcel | Q02248 | Mouse |
| Elongation factor 1-alpha 1 | 234 | c->w | ldcil | P10126 | Mouse |
| Myelin proteolipid protein | 220 | c->y | kvcgs | P60202 | Mouse |
| E3 ubiquitin-protein ligase XIAP | 90 | c->y | pncrf | A2BGY6 | Mouse |
Table 3.
Functional analysis of predicted cysteine S-nitrosylation sites with mutations.
| Analysis | Functional object | Observed | Expected | +/− | p value |
|---|---|---|---|---|---|
| PANTHER Pathways | Blood Coagulation | 13 | 3.82 | + | 3.64E-29 |
| PANTHER Protein Classification | Receptor (includes G-protein coupled receptor) | 190 | 123.24 | + | 4.13E-07 |
| Cytoskeletal protein | 98 | 64.03 | + | 5.46E-03 | |
| Defense/immunity protein | 72 | 46.53 | + | 4.48E-02 | |
| GO Biological Process | Muscle contraction | 73 | 37.48 | + | 1.92E-05 |
| Neurological system process | 188 | 128.53 | + | 2.04E-05 | |
| Cellular component organization | 131 | 84.15 | + | 9.63E-05 | |
| GO Molecular Function | Receptor activity (includes G-protein coupled receptor activity) | 190 | 124.18 | + | 6.02E-07 |
| Motor activity | 26 | 9.52 | + | 1.05E-03 | |
| Structural constituent of cytoskeleton | 98 | 64.03 | + | 4.52E-03 | |
| GO Cellular Component | Cytoskeleton | 98 | 64.03 | + | 1.09E-03 |
| Microtubule | 35 | 17.70 | + | 5.89E-03 | |
| Intracellular | 107 | 76.44 | + | 1.36E-02 |
3. Results and Discussion
Table S1 is a resource for researchers or clinicians to quickly identify SNVs associated with arrhythmias. The inclusion of information such as flanking base pairs and exact chromosomal position for each of the variants in Table S1 expedites the process of testing patients if whole genome, targeted sequencing data or proteomics data is available. Below are details of some of the genes and variations associated with them that correlate to arrhythmia.
3.1. NOS1AP
As shown in Table 1, the gene with the most variations was NOS1AP. The NOS1AP gene is located on chromosome 1 at q23.3. The gene codes for a regulatory protein—carboxyl-terminal PDZ ligand of neuronal nitric oxide synthase [32]. The protein influences the activity of nitric oxide synthase (NOS). When the NOS1AP protein is expressed in the heart, cardiac repolarization is typically accelerated. The NOS1AP enzyme inhibits l-type calcium channels [33]. The closing of l-type calcium channels inhibits the influx of Ca2+ current. Consequently, intracellular calcium concentrations do not increase, and the beta-adrenoreceptor stimulation of the heart is suppressed. This system, starting with the expression of the NOS1AP protein and resulting in a change in the electrophysiology of the heart, helps explain the role of NOS1AP variants with QT interval duration. QT interval is a measure of cardiac repolarization and a common biomarker of arrhythmia. The QT interval is estimated to be 30% heritable [34]. Studies have shown that the noncoding variants in NOS1AP are influential [35]. This is consistent with the data in Table 1 where all of the NOS1AP variants are intronic. The genome-wide association study by Arking et al. investigated 200 individuals and found associations in these individuals between QT interval length and the common variants in the noncoding regions of NOS1AP. These findings were replicated in a community of Old Order Amish [36].
Due to the complexity of the biological system, it is difficult to predict how these intronic variants specifically influence the malfunction or mis-regulation of the NOS1AP gene. However, the presence of resources such as Table S1 can help organize further inquiries into the role of intronic variants in disease development.
3.2. KCNN3
The SNV rs1131820 is a synonymous SNV found in the KCNN3 gene. The KCNN3 gene is located on chromosome 1q21 and is responsible for coding the protein SK3. The protein SK3 belongs to a family of proteins that operate as calcium activated potassium channels. Specifically, SK3 is a small-conductance calcium activated potassium channel [37]. This indicates that SK3 is voltage insensitive and the opening of the channel is instead reliant on the presence of calcium ions. The protein plays a role after hyper-polarization, a calcium dependent process that is executed after the firing of an action potential in a neuron. A variant in KCNN3 may influence the regulation or function of the SK3 protein. Reduced efficacy of the SK3 protein can inhibit influx of potassium ions through the potassium channel across a membrane. This can disrupt firing of the neuron. Consequently, KCNN3 variants expressed in regions such as the brain or the heart can have significant consequences on those delicate biological systems.
In the heart, a KCNN3 variant was shown to have an association with lone atrial fibrillation. This SNP rs1131820, shown in Table 1 is synonymous. This indicates that there is no consequent change in amino acid from the base pair mutation. One would anticipate that without a change in the amino acid, the effects of a variation would not manifest. However, in this circumstance, a carrier of the two major alleles GG at rs1131820 had an odds ratio of 2.85 (95% CI 1.13–7.18, p = 0.026) for lone atrial fibrillation when compared to carriers of the minor allele AA [38]. This suggests that carriers of the two major alleles GG are nearly three times more likely to develop lone atrial fibrillation. It is still unclear how a synonymous SNP can drastically increase the likelihood of an individual developing a disease or condition. Recent research has shown that these silent variations may affect the affinity of RNA binding proteins for mRNA, the splicing of pre-mRNA, and the stability of pre-mRNA [39,40,41]. This, in turn, may compromise the structure of the mRNA, and consequently influence protein formation, efficacy or concentration [42,43]. Once protein function or concentration has been altered, action potential firing in the heart can be disrupted and lead to the development of atrial fibrillation.
3.3. Analysis of Exonic SNVs
The exonic SNVs shown in Table 1 were explored further using the NHLBI Exome Sequencing Project (ESP). This project is a collaborative effort between various institutions and research hospitals to use NGS of the human exome to discover new genes and the mechanisms through which they influence the development of heart, blood, and lung disorders. The project expands across diverse populations and facilitates the sharing of conclusions and datasets throughout the scientific community (Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA, USA [44]. 11 exonic SNVs in the ESP database were searched. The new noteworthy information that the ESP database yielded for each SNV included variation impact analysis values. This value helps predict the likely impact of an amino acid substitution on protein structure and function [45]. The PolyPhen-2 scores for the variants ranged from benign 0.0 for the SNVs rs6795970, rs180514, rs1805127, rs12621643, and rs1800172—to possibly damaging 0.952 for SNV rs1805123. A score of benign suggests that the amino acid substitutions in each of these SNPs have no serious consequences on protein function. However, rs679570 was associated with time changes in the electrical cycles of the heart and an increased risk of heart block [46]. Conversely, rs846111 was associated with changes in the Q-T interval of the heart [47]. Finally, although rs113180 was not assigned a PolyPhen-2 score, this synonymous SNP is associated with a nearly three times higher likelihood of an individual developing lone atrial fibrillation [38].
3.4. Non-Synonymous Variation
Table 1 shows ten nsSNVs, one is a variant in a gene that encodes a ring finger protein, the other nine SNVs are found in genes that encode sodium ion channels or potassium voltage-gated channels. The two sodium ion channel genes are SCN10A and SCN5A. They are the type X and type V voltage-gated sodium channels, respectively. Table 1 shows that the SCN5A gene hosts two nsSNVs. The first is an amino acid substitution from serine to tyrosine at position 1103. The second is an amino acid substitution from histidine to arginine at position 558. The gene SCN10A contains the SNV rs6795970, an A/G base pair polymorphism at chromosome 3 position 38766675. This mutation in the exonic region results in an amino acid change from valine to alanine at position 1073. The specific voltage-gated sodium channel is labeled Nav1.8 and the protein is more commonly known for its role in facilitating cold perception in afferent nociceptive fibers [1]. Despite the association of this SNV with cardiac arrhythmias, the role of the Nav1.8 sodium channel in the electrophysiology of the heart is still uncharacterized [2]. The influence of the amino acid substitution on the function of the voltage-gated sodium channel is still unknown.
The genes found with nsSNVs that code for potassium voltage-gated channels are KCNE1, KCNH2, KCNQ1, and KCNE4. KCNH2 and KCNQ1, found in chromosome 7 and 11 respectively, are genes whose variants have been associated with long QT syndrome [48,49]. In KCNH2, the two nsSNVs are substitutions from lysine to threonine at amino acid positions 897 and 557. In KCNQ1, the variant results in an amino acid substitution from glycine to serine at position 643. The final two genes, KCNE1 and KCNE4 are found on chromosome 21 and 2, respectively. They belong to the KCNE family of genes, which encode trans-membrane proteins with a wide array of functions. Each of these genes contains non-synonymous SNPs that have associations with cardiac arrhythmias [50,51].
The RNF207 gene codes for a ring finger protein and contains the variant rs846111. This variant is found on chromosome 1 position 6279370. RNF207 codes for ring finger protein 207. The protein plays a role in intracellular zinc ion binding [3]. The specific in vivo function of the protein is still unknown. Although it is known that the variation translates to a shift of glycine to alanine at position 603, the specific influences of this amino acid substitution on the protein’s function are not yet understood.
3.5. Nitrosylation
PTMs such as glycosylation, phosphorylation, and nitrosylation help maintain cardiac rhythm, and modulate gating, localization, and cardiac channel expression levels [12]. To identify additional variations potentially connected to cardiac arrhythmias, we investigated the PTM Nitrosylation. Nitric oxide (NO) is synthesized by a majority of cardiac cell types and plays a vital role in regulating cardiac function [52]. The effects of NO are moderated by S-nitrosylation, which is the covalent modification of a protein cysteine thiol by an NO group to generate an S-nitrosothiol (SNO) [53]. The fundamental roles for S-nitrosylation have been involved in major functions of NO in the cardiovascular system [53].
Gould et al. performed a literature search for S-nitrosylated proteins in vivo [27]. The 233 S-nitrosylated proteins they compiled display an over-representation of mitochondrial proteins, a significant proportion involved in the generation of precursor metabolites and energy, and pathological conditions associated with an overproduction of NO, which results in inappropriate S-nitroyslation and dysfunction of proteins [27]. To further understand which nitrosylated proteins were affected by variation, we mapped the nitrosylation sites of 233 S-nitrosylated proteins to non-synonymous variations. We found seven cysteines that have loss of S-nitrosylation sites due to nsSNVs (Table 2).
Myosin, a motor protein listed in Table 2, plays a role in regulation of the heartbeat and cardiac function. Studies have shown that mutations of the cardiac myosin gene are the most common cause of inherited hypertrophic cardiomyopathy [54]. Nearly 200 disease-associated myosin mutations have been identified since the discovery of the β cardiac myosin heavy-chain (β-MHC) gene missense mutation’s role in hypertrophic cardiomyopathy [54,55]. β-MHC is a sarcomeric protein expressed in the right and left ventricles of the heart, as well as many skeletal muscles [56]. The precise location of the mutation or alteration of the myosin gene appears to influence the survival of patients; further investigation of this variation may lead to better prognostics for people affected by the mutation [55].
The current list of experimentally verified nitrosylation sites is not comprehensive. Table S2 contains the list of 1429 proteins which have conserved cysteines across eukaryotic species predicted to be S-nitrosylated and affected by nsSNPs and Figure 1 shows that several of these sites have nsSNVs. To better understand the distribution of predicted S-nitroyslation sites with conserved motifs, we analyzed the dataset to observe if there was over- or under-representation (compared to the complete human proteome) of pathways and functional elements/biological processes. An initial analysis using the UniProtKB/Swiss-Prot keyword ‘Disease’ showed that the keyword was over-represented in the predicted list of S-nitrosylated proteins (observed: 356; expected: 196.96; p-value: 3.64E-29). The highly significant p value proved that there are a higher number of disease proteins that are present in Table S2. For several of the known or predicted S-nitrosylation sites that are lost due to nsSNVs we were able to identify diseases associated with these variations (examples—dbSNP id: rs104894204, gene: CSRP3, UniProt AC/position/variation: P50461/58/c->g, disease: familial hypertrophic cardiomyopathy type 12; dbSNP id: rs121909300, gene: OXCT1, UniProt AC/position/variation: P55809/456/c->f, disease: succinyl-coa-3-ketoacid-coa transferase deficiency; dbSNP id: rs121909267, gene: CASR, UniProt AC/position/variation: P41180/131/c->w, disease: familial isolated hypoparathyroidism). S-nitrosylation has not yet been directly connected to these diseases and hence sites such as these (additional ones available in Table S2) are ideal for further biochemical analysis to elucidate the role of S-nitrosylation in cardiac and other diseases.
The PANTHER gene ontology database [57] determined if the predicted loss of S-nitrosylation sites affected by nsSNVs were statistically enriched with a particular molecular function or biological process. Over- and under-representation of PANTHER Pathways, Protein Classification, and Gene Ontology (GO) terms in the predicted list of mutated cysteines, provided an overview of possible effects these non-synonymous variations might have. Table 3 contains the results of the PANTHER statistical overrepresentation test. The major pathway identified as over-represented is blood coagulation (observed: 13; expected: 3.82; p-value: 3.64E-29). Atrial Fibrillation, a common type of arrhythmia, can lead to blood clots and other complications. Antithrombotic prophylaxis for stroke, a major risk of atrial fibrillation, is associated with an increased risk of bleeding [58].
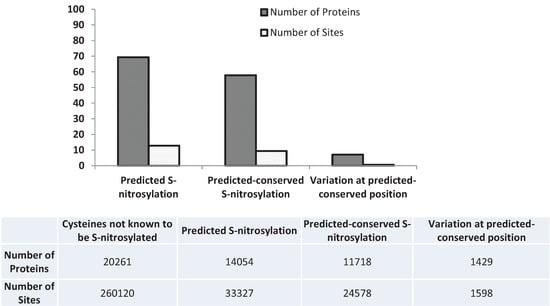
Figure 1.
Percentage of predicted S-nitrosylated proteins and sites and the ones which are conserved across mouse, fly, plant or yeast and the nsSNVs mapped to these proteins and sites. Details are available in Table S2.
The major broad terms for Gene Ontology (GO) Biological Processes that were statistically over-represented were Muscle Contraction, Neurological System Process, and Cellular Organization. For GO Molecular Function, the major terms included receptor activity (includes G-protein coupled receptor activity), motor activity, and structural constituent of cytoskeleton. The cytoskeleton was the top over-represented GO cellular component (p-value: 1.09E-03). The cytoskeleton of cardiac myocytes consists of actin filaments, intermediate filaments, and alpha- and beta-tubulin that form the microtubules by polymerization [59]. Studies have shown that the extra-sarcomeric cytoskeleton plays a part in the growth response of the heart and in the pathogenesis of cardiomyopathies [60]. Acquired forms of heart failure have displayed an altered expression of cytoskeletal proteins [61].
Microtubules were also statistically over-represented in GO cellular components (p-value: 5.89E-03). Recent studies have investigated the role of microtubules in cardiac arrhythmias. In hypertrophy and heart failure, the accumulation of microtubules disrupts sarcomere motion, contributing to declining ventricular compliance [61]. Microtubule integrity has been linked to cardio-protection, while microtubule disruption has been associated in the response to ischemia in cardiac myocytes [62].
3.6. Next-Generation Sequencing and Variation
As the cost of exome sequencing decreases, the use of patient exome sequencing will increase as a diagnostic tool. Here we describe a possible workflow for identifying variation from NGS data using a High-performance Integrated Virtual Environment (HIVE), a cloud-based environment [63].
The FASTA sequences surrounding a SNV are obtained from dbSNP by querying the database using the dnSNP ID from Table S1 followed by “Send to” option and selecting “File” at “Choose Destination” ≥ “FASTA”. This file is then uploaded to HIVE and used as a reference to which NGS data from an individual can be mapped (for additional details see help section on HIVE). Once the input files (FASTA file downloaded from dbSNP and NGS data obtained from an individual) are selected then HIVE-hexagon, a sequence mapping algorithm, is used with default parameters to map the NGS reads to the FASTA record which has the SNV in it. Once the mapping is complete then HIVE-pentagon, a SNV profiling tool, is used to identify all the variations in the individual. Visualization of the mapping results provides an easy overview of the variations present in the individual an example of which is shown in Figure 2.
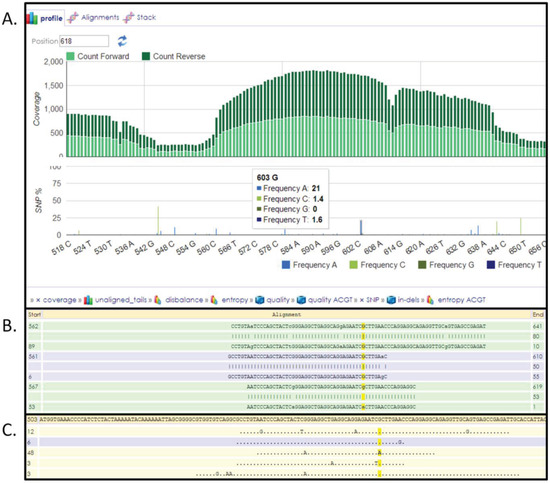
Figure 2.
HIVE interface showing results obtained from SNV profiling of human exome reads mapped to FASTA sequence surrounding a SNV. (A) Overall coverage result with the 603 position showing variation. (B) Reads mapped to the reference with the yellow highlighting the column selected. (C) Only variations are shown in this panel.
4. Conclusions
We expect that as whole genome sequencing becomes more affordable, genetic testing will play a larger role in the diagnosis of cardiac disorders, and that resources such as Table S1 will become more valuable and common place. In addition to their capacity to sequence whole genomes, the NGS platforms are capable of targeted sequencing. Table S1 also contains nucleotides that are upstream and downstream of each variant. This information allows researchers or physicians to quickly generate libraries of nucleic acids as the primary step in creation of a template. There is currently no quick and easy way to compile a list of variations that have associations with a condition or disease. This table provides a solution for this gap through manual curation. Rather than having physicians and researchers sift through papers to isolate SNPs that are associated with diseases, they can simply create a library of nucleic acids using the information contained in the table. These libraries can then be clonally amplified and their fragments can be prepared specifically per the requirements of the various NGS platforms. The list of these variations is also available from the BioMuta database [64]. It is important to note that DNA variants are rarely the sole determinant of whether an individual will acquire a disease or not. Consequently, it is important that healthcare workers as well as patients remember that these SNVs are a single parameter in a complex biological system. An additional danger that arises from sequencing patients as a diagnostic tool is the incidental discovery of presumed deleterious mutations [65].
In an attempt to identify more variants potentially connected to the disease, we investigated the post-translational modification nitrosylation in depth. Many studies have examined potential associations between acquired arrhythmias and post-translational modifications, such as glycosylation, phosphorylation, and nitrosylation. Table 2 and Table 3 provide both experimental and predicted approaches for discovering loss of S-nitrosylation sites that are affected by nsSNVs and details of the amino acid changes are present in Table S2. This information can be used to develop proteomic strategies for variation detection.
Supplementary Files
Acknowledgments
This project is supported in part by Research Participation Program at the Center for Biologics Evaluation and Research administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the U.S. Food and Drug Administration.
Author Contributions
Conceived and designed the study: Raja Mazumder. Performed the study: Ayman Abunimer and Phuc Lam. Analyzed the data: Ayman Abunimer, Krista Smith, Tsung-Jung Wu, and Phuc Lam. Contributed tools: Vahan Simonyan. Wrote the paper: Ayman Abunimer and Krista Smith.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Darbar, D. Genomics, heart failure and sudden cardiac death. Heart Fail. Rev. 2010, 15, 229–238. [Google Scholar] [CrossRef]
- Mahida, S.; Lubitz, S.A.; Rienstra, M.; Milan, D.J.; Ellinor, P.T. Monogenic atrial fibrillation as pathophysiological paradigms. Cardiovasc. Res. 2011, 89, 692–700. [Google Scholar] [CrossRef]
- Parvez, B.; Darbar, D. The “missing” link in atrial fibrillation heritability. J. Electrocardiol. 2011, 44, 641–644. [Google Scholar] [CrossRef]
- Biesecker, L.G. Opportunities and challenges for the integration of massively parallel genomic sequencing into clinical practice: Lessons from the clinseq project. Genet. Med. 2012, 14, 393–398. [Google Scholar] [CrossRef]
- Ng, D.; Johnston, J.J.; Teer, J.K.; Singh, L.N.; Peller, L.C.; Wynter, J.S.; Lewis, K.L.; Cooper, D.N.; Stenson, P.D.; Mullikin, J.C.; et al. Interpreting secondary cardiac disease variants in an exome cohort. Circ. Cardiovasc. Genet. 2013, 6, 337–346. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef]
- Gonzaga-Jauregui, C.; Lupski, J.R.; Gibbs, R.A. Human genome sequencing in health and disease. Annu. Rev. Med. 2012, 63, 35–61. [Google Scholar] [CrossRef]
- Moore, B.; Hu, H.; Singleton, M.; de la Vega, F.M.; Reese, M.G.; Yandell, M. Global analysis of disease-related DNA sequence variation in 10 healthy individuals: Implications for whole genome-based clinical diagnostics. Genet. Med. 2011, 13, 210–217. [Google Scholar]
- Priori, S.G.; Barhanin, J.; Hauer, R.N.; Haverkamp, W.; Jongsma, H.J.; Kleber, A.G.; McKenna, W.J.; Roden, D.M.; Rudy, Y.; Schwartz, K.; et al. Genetic and molecular basis of cardiac arrhythmias: Impact on clinical management parts I and II. Circulation 1999, 99, 518–528. [Google Scholar] [CrossRef]
- Camm, A.J.; Janse, M.J.; Roden, D.M.; Rosen, M.R.; Cinca, J.; Cobbe, S.M. Congenital and acquired long qt syndrome. Eur. Heart J. 2000, 21, 1232–1237. [Google Scholar] [CrossRef]
- Marban, E. Cardiac channelopathies. Nature 2002, 415, 213–218. [Google Scholar] [CrossRef]
- Jagu, B.; Charpentier, F.; Toumaniantz, G. Identifying potential functional impact of mutations and polymorphisms: Linking heart failure, increased risk of arrhythmias and sudden cardiac death. Front. Physiol. 2013, 4, 254. [Google Scholar]
- Noble, D. Modeling the heart—From genes to cells to the whole organ. Science 2002, 295, 1678–1682. [Google Scholar] [CrossRef]
- Mayne, J.; Starr, A.E.; Ning, Z.; Chen, R.; Chiang, C.K.; Figeys, D. Fine tuning of proteomic technologies to improve biological findings: Advancements in 2011–2013. Anal. Chem. 2014, 86, 175–195. [Google Scholar]
- Li, J.; Su, Z.; Ma, Z.Q.; Slebos, R.J.; Halvey, P.; Tabb, D.L.; Liebler, D.C.; Pao, W.; Zhang, B. A bioinformatics workflow for variant peptide detection in shotgun proteomics. Mol. Cell. Proteomics 2011. [Google Scholar] [CrossRef]
- Butter, F.; Davison, L.; Viturawong, T.; Scheibe, M.; Vermeulen, M.; Todd, J.A.; Mann, M. Proteome-wide analysis of disease-associated snps that show allele-specific transcription factor binding. PLoS Genet. 2012, 8, e1002982. [Google Scholar] [CrossRef]
- Eisenberg, E.; Adamsky, K.; Cohen, L.; Amariglio, N.; Hirshberg, A.; Rechavi, G.; Levanon, E.Y. Identification of rna editing sites in the snp database. Nucleic Acids Res. 2005, 33, 4612–4617. [Google Scholar] [CrossRef]
- Bunger, M.K.; Cargile, B.J.; Sevinsky, J.R.; Deyanova, E.; Yates, N.A.; Hendrickson, R.C.; Stephenson, J.L., Jr. Detection and validation of non-synonymous coding snps from orthogonal analysis of shotgun proteomics data. J. Proteome Res. 2007, 6, 2331–2340. [Google Scholar] [CrossRef]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nature Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Bian, K.; Doursout, M.F.; Murad, F. Vascular system: Role of nitric oxide in cardiovascular diseases. J. Clin. Hypertens. 2008, 10, 304–310. [Google Scholar] [CrossRef]
- Greco, T.M.; Hodara, R.; Parastatidis, I.; Heijnen, H.F.; Dennehy, M.K.; Liebler, D.C.; Ischiropoulos, H. Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc. Nat. Acad. Sci. USA 2006, 103, 7420–7425. [Google Scholar] [CrossRef]
- Massy, Z.A.; Fumeron, C.; Borderie, D.; Tuppin, P.; Nguyen-Khoa, T.; Benoit, M.O.; Jacquot, C.; Buisson, C.; Drueke, T.B.; Ekindjian, O.G.; et al. Increased pasma S-nitrosothiol concentrations predict cardiovascular outcomes among patients with end-stage renal disease: A prospective study. J. Am. Soc. Nephrol. 2004, 15, 470–476. [Google Scholar] [CrossRef]
- Datta, B.; Tufnell-Barrett, T.; Bleasdale, R.A.; Jones, C.J.; Beeton, I.; Paul, V.; Frenneaux, M.; James, P. Red blood cell nitric oxide as an endocrine vasoregulator: A potential role in congestive heart failure. Circulation 2004, 109, 1339–1342. [Google Scholar] [CrossRef]
- Herren, A.W.; Bers, D.M.; Grandi, E. Post-translational modifications of the cardiac na channel: Contribution of camkii-dependent phosphorylation to acquired arrhythmias. American J. Physiol. Heart Circ. Physiol. 2013, 305, H431–H445. [Google Scholar] [CrossRef]
- Cutler, M.J.; Plummer, B.N.; Wan, X.; Sun, Q.A.; Hess, D.; Liu, H.; Deschenes, I.; Rosenbaum, D.S.; Stamler, J.S.; Laurita, K.R. Aberrant s-nitrosylation mediates calcium-triggered ventricular arrhythmia in the intact heart. Proc. Nat. Acad. Sci. USA 2012, 109, 18186–18191. [Google Scholar] [CrossRef]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Federhen, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2012, 40, D13–D25. [Google Scholar] [CrossRef]
- Gould, N.; Doulias, P.T.; Tenopoulou, M.; Raju, K.; Ischiropoulos, H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J. Biol. Chem 2013, 288, 26473–26479. [Google Scholar] [CrossRef]
- Xue, Y.; Liu, Z.; Gao, X.; Jin, C.; Wen, L.; Yao, X.; Ren, J. Gps-sno: Computational prediction of protein S-nitrosylation sites with a modified gps algorithm. PLoS One 2010, 5, e11290. [Google Scholar]
- Karagiannis, K.; Simonyan, V.; Mazumder, R. Snvdis: A proteome-wide analysis service for evaluating nssnvs in protein functional sites and pathways. Genomics Proteomics Bioinformatics 2013, 11, 122–126. [Google Scholar] [CrossRef]
- Mazumder, R.; Morampudi, K.S.; Motwani, M.; Vasudevan, S.; Goldman, R. Proteome-wide analysis of single-nucleotide variations in the n-glycosylation sequon of human genes. PLoS One 2012, 7, e36212. [Google Scholar]
- Mi, H.; Thomas, P. Panther pathway: An ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 2009, 563, 123–140. [Google Scholar] [CrossRef]
- Earle, N.; Yeo Han, D.; Pilbrow, A.; Crawford, J.; Smith, W.; Shelling, A.N.; Cameron, V.; Love, D.R.; Skinner, J.R. Single nucleotide polymorphisms in arrhythmia genes modify the risk of cardiac events and sudden death in long qt syndrome. Heart Rhythm 2013, 11, 76–82. [Google Scholar]
- Chang, K.C.; Barth, A.S.; Sasano, T.; Kizana, E.; Kashiwakura, Y.; Zhang, Y.; Foster, D.B.; Marban, E. Capon modulates cardiac repolarization via neuronal nitric oxide synthase signaling in the heart. Proc. Nat. Acad. Sci. USA 2008, 105, 4477–4482. [Google Scholar] [CrossRef]
- Arking, D.E.; Pfeufer, A.; Post, W.; Kao, W.H.; Newton-Cheh, C.; Ikeda, M.; West, K.; Kashuk, C.; Akyol, M.; Perz, S.; et al. A common genetic variant in the nos1 regulator nos1ap modulates cardiac repolarization. Nat. Genet. 2006, 38, 644–651. [Google Scholar] [CrossRef]
- Wratten, N.S.; Memoli, H.; Huang, Y.; Dulencin, A.M.; Matteson, P.G.; Cornacchia, M.A.; Azaro, M.A.; Messenger, J.; Hayter, J.E.; Bassett, A.S.; et al. Identification of a schizophrenia-associated functional noncoding variant in nos1ap. Am. J. Psychiatry 2009, 166, 434–441. [Google Scholar] [CrossRef]
- Post, W.; Shen, H.; Damcott, C.; Arking, D.E.; Kao, W.H.; Sack, P.A.; Ryan, K.A.; Chakravarti, A.; Mitchell, B.D.; Shuldiner, A.R. Associations between genetic variants in the nos1ap (capon) gene and cardiac repolarization in the old order amish. Hum. Hered. 2007, 64, 214–219. [Google Scholar] [CrossRef]
- Ellinor, P.T.; Lunetta, K.L.; Glazer, N.L.; Pfeufer, A.; Alonso, A.; Chung, M.K.; Sinner, M.F.; de Bakker, P.I.; Mueller, M.; Lubitz, S.A.; et al. Common variants in kcnn3 are associated with lone atrial fibrillation. Nat. Genet. 2010, 42, 240–244. [Google Scholar] [CrossRef]
- Olesen, M.S.; Jabbari, J.; Holst, A.G.; Nielsen, J.B.; Steinbruchel, D.A.; Jespersen, T.; Haunso, S.; Svendsen, J.H. Screening of kcnn3 in patients with early-onset lone atrial fibrillation. Europace 2011, 13, 963–967. [Google Scholar] [CrossRef]
- Sauna, Z.E.; Kimchi-Sarfaty, C.; Ambudkar, S.V.; Gottesman, M.M. The sounds of silence: Synonymous mutations affect function. Pharmacogenomics 2007, 8, 527–532. [Google Scholar] [CrossRef]
- Hunt, R.; Sauna, Z.E.; Ambudkar, S.V.; Gottesman, M.M.; Kimchi-Sarfaty, C. Silent (synonymous) snps: Should we care about them? Methods Mol. Biol. 2009, 578, 23–39. [Google Scholar] [CrossRef]
- Chamary, J.V.; Parmley, J.L.; Hurst, L.D. Hearing silence: Non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 2006, 7, 98–108. [Google Scholar] [CrossRef]
- Purvis, I.J.; Bettany, A.J.; Santiago, T.C.; Coggins, J.R.; Duncan, K.; Eason, R.; Brown, A.J. The efficiency of folding of some proteins is increased by controlled rates of translation in vivo. A hypothesis. J. Mol. Biol. 1987, 193, 413–417. [Google Scholar] [CrossRef]
- Nackley, A.G.; Shabalina, S.A.; Tchivileva, I.E.; Satterfield, K.; Korchynskyi, O.; Makarov, S.S.; Maixner, W.; Diatchenko, L. Human catechol-o-methyltransferase haplotypes modulate protein expression by altering mrna secondary structure. Science 2006, 314, 1930–1933. [Google Scholar] [CrossRef]
- HLBI_Exome_Sequencing_Project. Exome Variant Server. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 9 January 2013).
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Chambers, J.C.; Zhao, J.; Terracciano, C.M.; Bezzina, C.R.; Zhang, W.; Kaba, R.; Navaratnarajah, M.; Lotlikar, A.; Sehmi, J.S.; Kooner, M.K.; et al. Genetic variation in scn10a influences cardiac conduction. Nat. Genet. 2010, 42, 149–152. [Google Scholar] [CrossRef]
- Pfeufer, A.; Sanna, S.; Arking, D.E.; Muller, M.; Gateva, V.; Fuchsberger, C.; Ehret, G.B.; Orru, M.; Pattaro, C.; Kottgen, A.; et al. Common variants at ten loci modulate the qt interval duration in the qtscd study. Nat. Genet. 2009, 41, 407–414. [Google Scholar] [CrossRef]
- Pfeufer, A.; Jalilzadeh, S.; Perz, S.; Mueller, J.C.; Hinterseer, M.; Illig, T.; Akyol, M.; Huth, C.; Schopfer-Wendels, A.; Kuch, B.; et al. Common variants in myocardial ion channel genes modify the qt interval in the general population: Results from the kora study. Circ. Res. 2005, 96, 693–701. [Google Scholar] [CrossRef]
- Ozawa, T.; Ito, M.; Tamaki, S.; Yao, T.; Ashihara, T.; Kita, Y.; Okamura, T.; Ueshima, H.; Horie, M. Gender and age effects on ventricular repolarization abnormality in japanese general carriers of a g643s common single nucleotide polymorphism for the kcnq1 gene. Circ. J. Offic. J. Jpn. Circ. Soc. 2006, 70, 645–650. [Google Scholar]
- Zeng, Z.Y.; Pu, J.L.; Tan, C.; Teng, S.Y.; Chen, J.H.; Su, S.Y.; Zhou, X.Y.; Zhang, S.; Li, Y.S.; Wang, F.Z.; et al. The association of single nucleotide polymorphism of slow delayed rectifier k+ channel genes with atrial fibrillation in han nationality chinese. Zhonghua Xin Xue Guan Bing Za Zhi 2005, 33, 987–991. [Google Scholar]
- Olszak-Waskiewicz, M.; Kubik, L.; Dziuk, M.; Sidlo, E.; Kucharczyk, K.; Kaczanowski, R. The association between scn5a, kcnq1 and kcne1 gene polymorphisms and complex ventricular arrhythmias in survivors of myocardial infarction. Kardiol. Pol. 2008, 66, 845–853. [Google Scholar]
- Tamargo, J.; Caballero, R.; Gomez, R.; Delpon, E. Cardiac electrophysiological effects of nitric oxide. Cardiovasc. Res. 2010, 87, 593–600. [Google Scholar] [CrossRef]
- Lima, B.; Forrester, M.T.; Hess, D.T.; Stamler, J.S. S-nitrosylation in cardiovascular signaling. Circ. Res. 2010, 106, 633–646. [Google Scholar] [CrossRef]
- Herron, T.J.; Devaney, E.J.; Metzger, J.M. Modulation of cardiac performance by motor protein gene transfer. Ann. N. Y. Acad. Sci. 2008, 1123, 96–104. [Google Scholar]
- Watkins, H.; Rosenzweig, A.; Hwang, D.S.; Levi, T.; McKenna, W.; Seidman, C.E.; Seidman, J.G. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N. Engl. J. Med. 1992, 326, 1108–1114. [Google Scholar] [CrossRef]
- Yu, Q.T.; Ifegwu, J.; Marian, A.J.; Mares, A., Jr.; Hill, R.; Perryman, M.B.; Bachinski, L.L.; Roberts, R. Hypertrophic cardiomyopathy mutation is expressed in messenger rna of skeletal as well as cardiac muscle. Circulation 1993, 87, 406–412. [Google Scholar] [CrossRef]
- Mi, H.; Lazareva-Ulitsky, B.; Loo, R.; Kejariwal, A.; Vandergriff, J.; Rabkin, S.; Guo, N.; Muruganujan, A.; Doremieux, O.; Campbell, M.J.; et al. The panther database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005, 33, D284–D288. [Google Scholar] [CrossRef]
- You, J.J.; Singer, D.E.; Howard, P.A.; Lane, D.A.; Eckman, M.H.; Fang, M.C.; Hylek, E.M.; Schulman, S.; Go, A.S.; Hughes, M.; et al. Antithrombotic therapy for atrial fibrillation: Antithrombotic therapy and prevention of thrombosis, 9th ed: American college of chest physicians evidence-based clinical practice guidelines. Chest 2012, 141, e531S–e575S. [Google Scholar]
- Hein, S.; Kostin, S.; Heling, A.; Maeno, Y.; Schaper, J. The role of the cytoskeleton in heart failure. Cardiovasc. Res. 2000, 45, 273–278. [Google Scholar] [CrossRef]
- Roberts, R.; Schwartz, K. Myocardial diseases. Circulation 2000, 102, IV34–IV39. [Google Scholar]
- Heling, A.; Zimmermann, R.; Kostin, S.; Maeno, Y.; Hein, S.; Devaux, B.; Bauer, E.; Klovekorn, W.P.; Schlepper, M.; Schaper, W.; et al. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ. Res. 2000, 86, 846–853. [Google Scholar] [CrossRef]
- Xiao, J.; Cao, H.; Liang, D.; Liu, Y.; Zhang, H.; Zhao, H.; Li, J.; Yan, B.; Peng, L.; Zhou, Z.; et al. Taxol, a microtubule stabilizer, prevents ischemic ventricular arrhythmias in rats. J. Cell. Mol. Med. 2011, 15, 1166–1176. [Google Scholar] [CrossRef]
- Simonyan, V.; Mazumder, R. High-performance integrated virtual environment clouds (hive) for extra-large (xl) data analysis. Comparative sequence, genome analysis, genome assembly, & genome scale computational methods session. In Proceesings of the 2011 International Conference on Bioinformatics and Computational Biology, Las Vegas, NV, USA, 17–21 July 2011.
- Wu, T.-J.; Shamsaddini, A.; Pan, Y.; Smith, K.; Crichton, D.; Simonyan, V.; Mazumder, R. A framework for organizing cancer related variations from existing databases, publications and ngs data using a high-performance integrated virtual environment (HIVE). Database 2014, in press. [Google Scholar]
- Johnston, J.J.; Rubinstein, W.S.; Facio, F.M.; Ng, D.; Singh, L.N.; Teer, J.K.; Mullikin, J.C.; Biesecker, L.G. Secondary variants in individuals undergoing exome sequencing: Screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibili ty genes. Am. J. Hum. Genet. 2012, 91, 97–108. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).