Single-Nucleotide Variations in Cardiac Arrhythmias: Prospects for Genomics and Proteomics Based Biomarker Discovery and Diagnostics

Abstract
:1. Introduction

{kind=link}
{kind=link}
| Gene | Number of Variations | Chromosome Number | Amino Acid Change | Gene Description |
|---|---|---|---|---|
| SCN10A | 2 | 3 | V1073A | Sodium ion channel |
| HEY2 | 1 | 6 | 0 | Cardiovascular helix-loop-helix factor 1 |
| SCN5A | 5 | 3 | S1103Y, H558R | Sodium ion channel |
| TBX5 | 1 | 12 | 0 | T-box transcription factor |
| NOS1AP | 12 | 1 | 0 | Nitric oxide synthase 1 adaptor protein |
| ATP1B1 | 2 | 1 | 0 | Sodium/potassium-transporting ATPase subunit beta-1 |
| ZFHX3 | 3 | 16 | 0 | Zinc finger homeobox protein 3 |
| KCNN3 | 3 | 1 | N44N | Small conductance potassium channel |
| KCNJ2 | 2 | 17 | 0 | Potassium ion channel |
| XYLB | 3 | 3 | 0 | Energy metabolism |
| EXOG | 3 | 3 | 0 | Endonuclease |
| ACVR2B | 3 | 3 | 0 | Activin Receptor |
| RNF207 | 1 | 1 | G603A | Ring finger protein |
| PLN | 2 | 6 | 0 | Cardiac Muscle |
| KCNH2 | 5 | 7 | K897T, K557T | Potassium voltage gated channel |
| KCNQ1 | 4 | 11 | G643S | Potassium voltage gated channel |
| LITAF | 1 | 16 | 0 | DNA binding protein |
| NDRG4 | 1 | 16 | 0 | Mitogenic signalling |
| AGTR1 | 1 | 3 | 0 | Angiotensis II receptor |
| KNG1 | 1 | 3 | 0 | Kininogen |
| KCNE1 | 3 | 21 | D85N, S38G | Potassium voltage gated channel |
| KCNE4 | 1 | 2 | D196E | Potassium voltage-gated channel |
2. Experimental
| Protein name | Position | Variation | Subseq | Ortholog | Species |
|---|---|---|---|---|---|
| Alpha-enolase | 357 | c->y | qackl | P21550 | Mouse |
| Cysteine and glycine-rich protein 3 | 58 | c->g | iyckv | P50462 | Mouse |
| Myosin-6 | 949 | c->y | decse | Q02566 | Mouse |
| Catenin beta-1 | 619 | c->y | vlcel | Q02248 | Mouse |
| Elongation factor 1-alpha 1 | 234 | c->w | ldcil | P10126 | Mouse |
| Myelin proteolipid protein | 220 | c->y | kvcgs | P60202 | Mouse |
| E3 ubiquitin-protein ligase XIAP | 90 | c->y | pncrf | A2BGY6 | Mouse |
| Analysis | Functional object | Observed | Expected | +/− | p value |
|---|---|---|---|---|---|
| PANTHER Pathways | Blood Coagulation | 13 | 3.82 | + | 3.64E-29 |
| PANTHER Protein Classification | Receptor (includes G-protein coupled receptor) | 190 | 123.24 | + | 4.13E-07 |
| Cytoskeletal protein | 98 | 64.03 | + | 5.46E-03 | |
| Defense/immunity protein | 72 | 46.53 | + | 4.48E-02 | |
| GO Biological Process | Muscle contraction | 73 | 37.48 | + | 1.92E-05 |
| Neurological system process | 188 | 128.53 | + | 2.04E-05 | |
| Cellular component organization | 131 | 84.15 | + | 9.63E-05 | |
| GO Molecular Function | Receptor activity (includes G-protein coupled receptor activity) | 190 | 124.18 | + | 6.02E-07 |
| Motor activity | 26 | 9.52 | + | 1.05E-03 | |
| Structural constituent of cytoskeleton | 98 | 64.03 | + | 4.52E-03 | |
| GO Cellular Component | Cytoskeleton | 98 | 64.03 | + | 1.09E-03 |
| Microtubule | 35 | 17.70 | + | 5.89E-03 | |
| Intracellular | 107 | 76.44 | + | 1.36E-02 |
3. Results and Discussion
3.1. NOS1AP
3.2. KCNN3
3.3. Analysis of Exonic SNVs
3.4. Non-Synonymous Variation
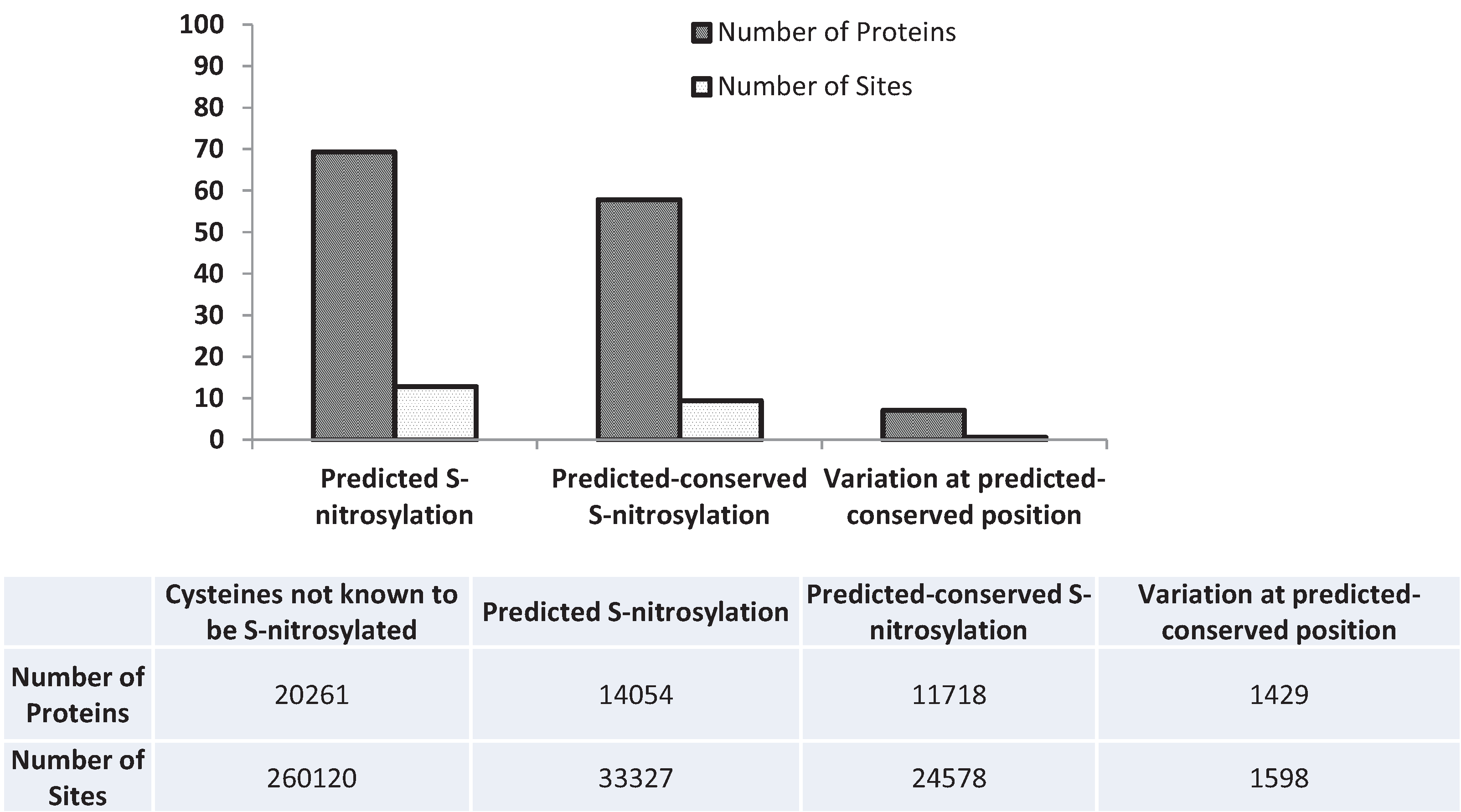
3.5. Nitrosylation
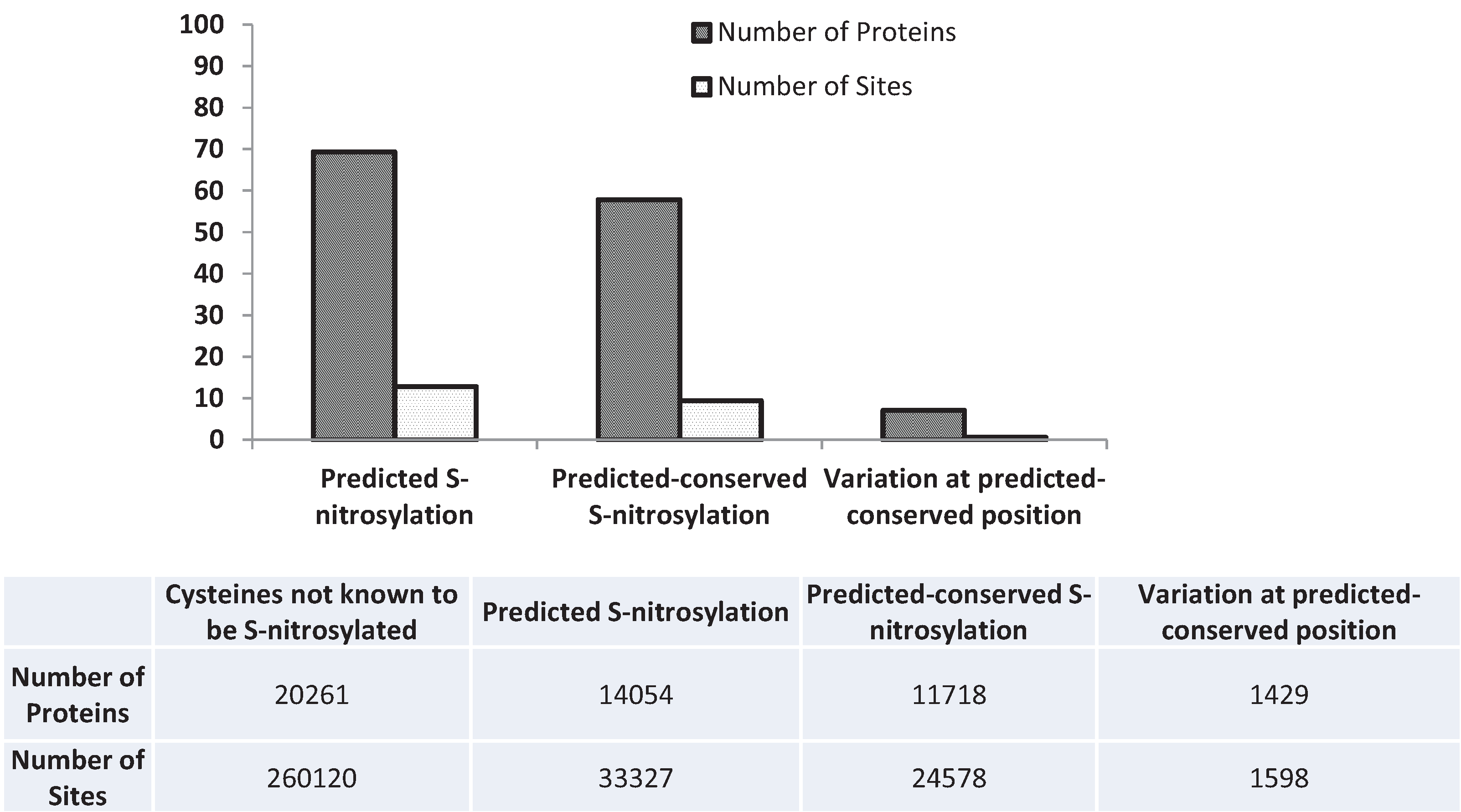
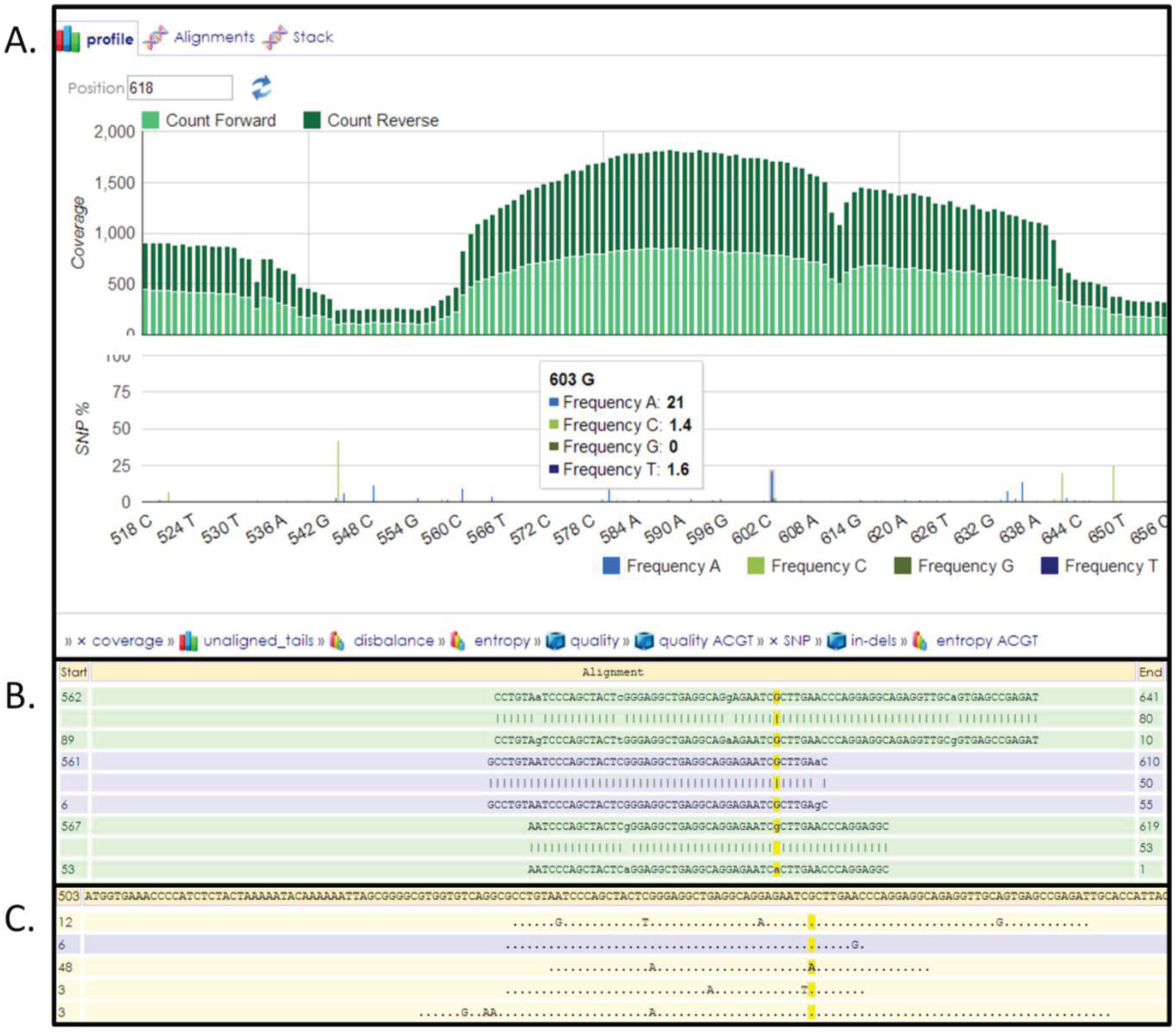
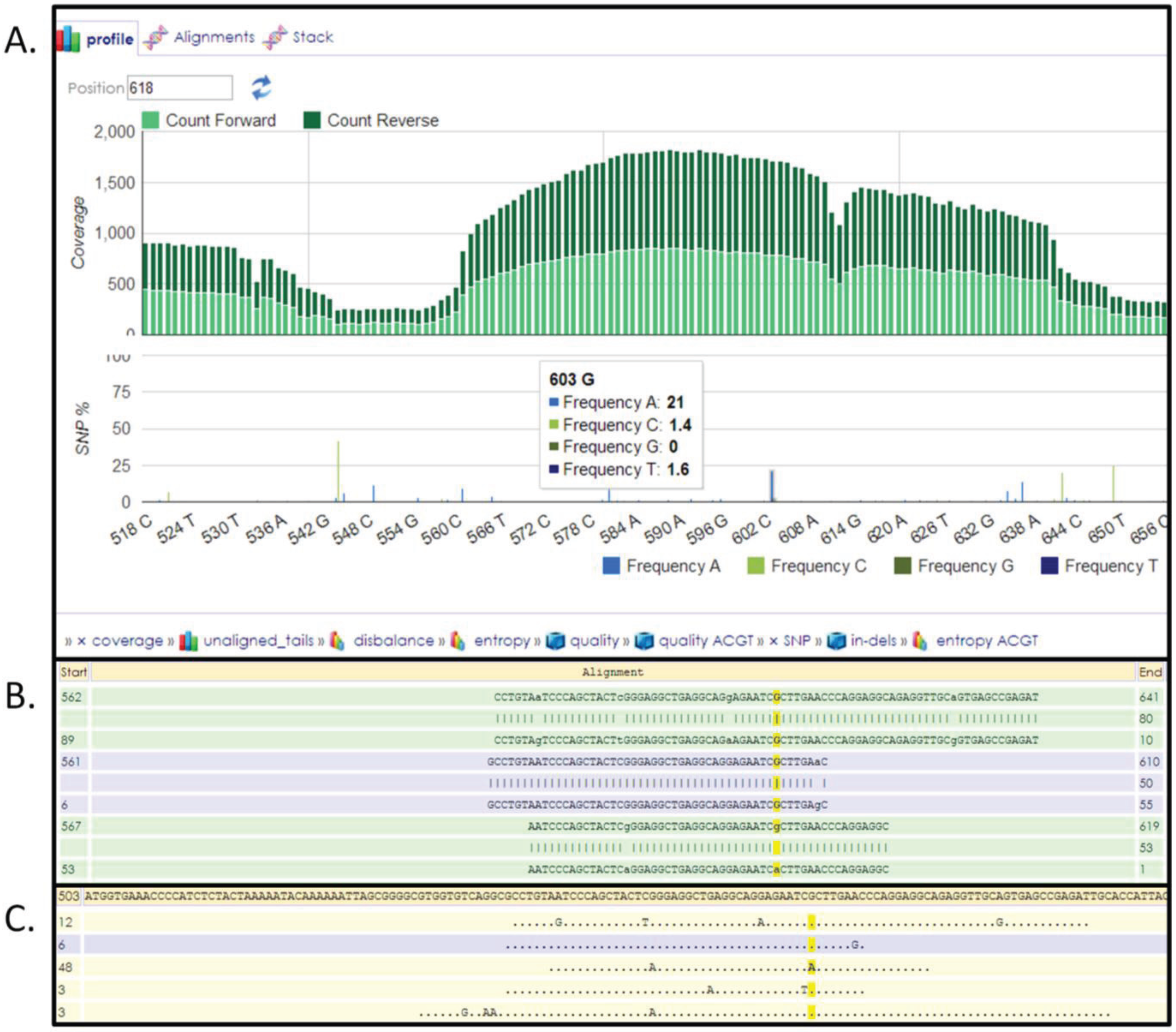
3.6. Next-Generation Sequencing and Variation
4. Conclusions
Supplementary Files
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Darbar, D. Genomics, heart failure and sudden cardiac death. Heart Fail. Rev. 2010, 15, 229–238. [Google Scholar] [CrossRef]
- Mahida, S.; Lubitz, S.A.; Rienstra, M.; Milan, D.J.; Ellinor, P.T. Monogenic atrial fibrillation as pathophysiological paradigms. Cardiovasc. Res. 2011, 89, 692–700. [Google Scholar] [CrossRef]
- Parvez, B.; Darbar, D. The “missing” link in atrial fibrillation heritability. J. Electrocardiol. 2011, 44, 641–644. [Google Scholar] [CrossRef]
- Biesecker, L.G. Opportunities and challenges for the integration of massively parallel genomic sequencing into clinical practice: Lessons from the clinseq project. Genet. Med. 2012, 14, 393–398. [Google Scholar] [CrossRef]
- Ng, D.; Johnston, J.J.; Teer, J.K.; Singh, L.N.; Peller, L.C.; Wynter, J.S.; Lewis, K.L.; Cooper, D.N.; Stenson, P.D.; Mullikin, J.C.; et al. Interpreting secondary cardiac disease variants in an exome cohort. Circ. Cardiovasc. Genet. 2013, 6, 337–346. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef]
- Gonzaga-Jauregui, C.; Lupski, J.R.; Gibbs, R.A. Human genome sequencing in health and disease. Annu. Rev. Med. 2012, 63, 35–61. [Google Scholar] [CrossRef]
- Moore, B.; Hu, H.; Singleton, M.; de la Vega, F.M.; Reese, M.G.; Yandell, M. Global analysis of disease-related DNA sequence variation in 10 healthy individuals: Implications for whole genome-based clinical diagnostics. Genet. Med. 2011, 13, 210–217. [Google Scholar]
- Priori, S.G.; Barhanin, J.; Hauer, R.N.; Haverkamp, W.; Jongsma, H.J.; Kleber, A.G.; McKenna, W.J.; Roden, D.M.; Rudy, Y.; Schwartz, K.; et al. Genetic and molecular basis of cardiac arrhythmias: Impact on clinical management parts I and II. Circulation 1999, 99, 518–528. [Google Scholar] [CrossRef]
- Camm, A.J.; Janse, M.J.; Roden, D.M.; Rosen, M.R.; Cinca, J.; Cobbe, S.M. Congenital and acquired long qt syndrome. Eur. Heart J. 2000, 21, 1232–1237. [Google Scholar] [CrossRef]
- Marban, E. Cardiac channelopathies. Nature 2002, 415, 213–218. [Google Scholar] [CrossRef]
- Jagu, B.; Charpentier, F.; Toumaniantz, G. Identifying potential functional impact of mutations and polymorphisms: Linking heart failure, increased risk of arrhythmias and sudden cardiac death. Front. Physiol. 2013, 4, 254. [Google Scholar]
- Noble, D. Modeling the heart—From genes to cells to the whole organ. Science 2002, 295, 1678–1682. [Google Scholar] [CrossRef]
- Mayne, J.; Starr, A.E.; Ning, Z.; Chen, R.; Chiang, C.K.; Figeys, D. Fine tuning of proteomic technologies to improve biological findings: Advancements in 2011–2013. Anal. Chem. 2014, 86, 175–195. [Google Scholar]
- Li, J.; Su, Z.; Ma, Z.Q.; Slebos, R.J.; Halvey, P.; Tabb, D.L.; Liebler, D.C.; Pao, W.; Zhang, B. A bioinformatics workflow for variant peptide detection in shotgun proteomics. Mol. Cell. Proteomics 2011. [Google Scholar] [CrossRef]
- Butter, F.; Davison, L.; Viturawong, T.; Scheibe, M.; Vermeulen, M.; Todd, J.A.; Mann, M. Proteome-wide analysis of disease-associated snps that show allele-specific transcription factor binding. PLoS Genet. 2012, 8, e1002982. [Google Scholar] [CrossRef]
- Eisenberg, E.; Adamsky, K.; Cohen, L.; Amariglio, N.; Hirshberg, A.; Rechavi, G.; Levanon, E.Y. Identification of rna editing sites in the snp database. Nucleic Acids Res. 2005, 33, 4612–4617. [Google Scholar] [CrossRef]
- Bunger, M.K.; Cargile, B.J.; Sevinsky, J.R.; Deyanova, E.; Yates, N.A.; Hendrickson, R.C.; Stephenson, J.L., Jr. Detection and validation of non-synonymous coding snps from orthogonal analysis of shotgun proteomics data. J. Proteome Res. 2007, 6, 2331–2340. [Google Scholar] [CrossRef]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nature Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Bian, K.; Doursout, M.F.; Murad, F. Vascular system: Role of nitric oxide in cardiovascular diseases. J. Clin. Hypertens. 2008, 10, 304–310. [Google Scholar] [CrossRef]
- Greco, T.M.; Hodara, R.; Parastatidis, I.; Heijnen, H.F.; Dennehy, M.K.; Liebler, D.C.; Ischiropoulos, H. Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc. Nat. Acad. Sci. USA 2006, 103, 7420–7425. [Google Scholar] [CrossRef]
- Massy, Z.A.; Fumeron, C.; Borderie, D.; Tuppin, P.; Nguyen-Khoa, T.; Benoit, M.O.; Jacquot, C.; Buisson, C.; Drueke, T.B.; Ekindjian, O.G.; et al. Increased pasma S-nitrosothiol concentrations predict cardiovascular outcomes among patients with end-stage renal disease: A prospective study. J. Am. Soc. Nephrol. 2004, 15, 470–476. [Google Scholar] [CrossRef]
- Datta, B.; Tufnell-Barrett, T.; Bleasdale, R.A.; Jones, C.J.; Beeton, I.; Paul, V.; Frenneaux, M.; James, P. Red blood cell nitric oxide as an endocrine vasoregulator: A potential role in congestive heart failure. Circulation 2004, 109, 1339–1342. [Google Scholar] [CrossRef]
- Herren, A.W.; Bers, D.M.; Grandi, E. Post-translational modifications of the cardiac na channel: Contribution of camkii-dependent phosphorylation to acquired arrhythmias. American J. Physiol. Heart Circ. Physiol. 2013, 305, H431–H445. [Google Scholar] [CrossRef]
- Cutler, M.J.; Plummer, B.N.; Wan, X.; Sun, Q.A.; Hess, D.; Liu, H.; Deschenes, I.; Rosenbaum, D.S.; Stamler, J.S.; Laurita, K.R. Aberrant s-nitrosylation mediates calcium-triggered ventricular arrhythmia in the intact heart. Proc. Nat. Acad. Sci. USA 2012, 109, 18186–18191. [Google Scholar] [CrossRef]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Federhen, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2012, 40, D13–D25. [Google Scholar] [CrossRef]
- Gould, N.; Doulias, P.T.; Tenopoulou, M.; Raju, K.; Ischiropoulos, H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J. Biol. Chem 2013, 288, 26473–26479. [Google Scholar] [CrossRef]
- Xue, Y.; Liu, Z.; Gao, X.; Jin, C.; Wen, L.; Yao, X.; Ren, J. Gps-sno: Computational prediction of protein S-nitrosylation sites with a modified gps algorithm. PLoS One 2010, 5, e11290. [Google Scholar]
- Karagiannis, K.; Simonyan, V.; Mazumder, R. Snvdis: A proteome-wide analysis service for evaluating nssnvs in protein functional sites and pathways. Genomics Proteomics Bioinformatics 2013, 11, 122–126. [Google Scholar] [CrossRef]
- Mazumder, R.; Morampudi, K.S.; Motwani, M.; Vasudevan, S.; Goldman, R. Proteome-wide analysis of single-nucleotide variations in the n-glycosylation sequon of human genes. PLoS One 2012, 7, e36212. [Google Scholar]
- Mi, H.; Thomas, P. Panther pathway: An ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 2009, 563, 123–140. [Google Scholar] [CrossRef]
- Earle, N.; Yeo Han, D.; Pilbrow, A.; Crawford, J.; Smith, W.; Shelling, A.N.; Cameron, V.; Love, D.R.; Skinner, J.R. Single nucleotide polymorphisms in arrhythmia genes modify the risk of cardiac events and sudden death in long qt syndrome. Heart Rhythm 2013, 11, 76–82. [Google Scholar]
- Chang, K.C.; Barth, A.S.; Sasano, T.; Kizana, E.; Kashiwakura, Y.; Zhang, Y.; Foster, D.B.; Marban, E. Capon modulates cardiac repolarization via neuronal nitric oxide synthase signaling in the heart. Proc. Nat. Acad. Sci. USA 2008, 105, 4477–4482. [Google Scholar] [CrossRef]
- Arking, D.E.; Pfeufer, A.; Post, W.; Kao, W.H.; Newton-Cheh, C.; Ikeda, M.; West, K.; Kashuk, C.; Akyol, M.; Perz, S.; et al. A common genetic variant in the nos1 regulator nos1ap modulates cardiac repolarization. Nat. Genet. 2006, 38, 644–651. [Google Scholar] [CrossRef]
- Wratten, N.S.; Memoli, H.; Huang, Y.; Dulencin, A.M.; Matteson, P.G.; Cornacchia, M.A.; Azaro, M.A.; Messenger, J.; Hayter, J.E.; Bassett, A.S.; et al. Identification of a schizophrenia-associated functional noncoding variant in nos1ap. Am. J. Psychiatry 2009, 166, 434–441. [Google Scholar] [CrossRef]
- Post, W.; Shen, H.; Damcott, C.; Arking, D.E.; Kao, W.H.; Sack, P.A.; Ryan, K.A.; Chakravarti, A.; Mitchell, B.D.; Shuldiner, A.R. Associations between genetic variants in the nos1ap (capon) gene and cardiac repolarization in the old order amish. Hum. Hered. 2007, 64, 214–219. [Google Scholar] [CrossRef]
- Ellinor, P.T.; Lunetta, K.L.; Glazer, N.L.; Pfeufer, A.; Alonso, A.; Chung, M.K.; Sinner, M.F.; de Bakker, P.I.; Mueller, M.; Lubitz, S.A.; et al. Common variants in kcnn3 are associated with lone atrial fibrillation. Nat. Genet. 2010, 42, 240–244. [Google Scholar] [CrossRef]
- Olesen, M.S.; Jabbari, J.; Holst, A.G.; Nielsen, J.B.; Steinbruchel, D.A.; Jespersen, T.; Haunso, S.; Svendsen, J.H. Screening of kcnn3 in patients with early-onset lone atrial fibrillation. Europace 2011, 13, 963–967. [Google Scholar] [CrossRef]
- Sauna, Z.E.; Kimchi-Sarfaty, C.; Ambudkar, S.V.; Gottesman, M.M. The sounds of silence: Synonymous mutations affect function. Pharmacogenomics 2007, 8, 527–532. [Google Scholar] [CrossRef]
- Hunt, R.; Sauna, Z.E.; Ambudkar, S.V.; Gottesman, M.M.; Kimchi-Sarfaty, C. Silent (synonymous) snps: Should we care about them? Methods Mol. Biol. 2009, 578, 23–39. [Google Scholar] [CrossRef]
- Chamary, J.V.; Parmley, J.L.; Hurst, L.D. Hearing silence: Non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 2006, 7, 98–108. [Google Scholar] [CrossRef]
- Purvis, I.J.; Bettany, A.J.; Santiago, T.C.; Coggins, J.R.; Duncan, K.; Eason, R.; Brown, A.J. The efficiency of folding of some proteins is increased by controlled rates of translation in vivo. A hypothesis. J. Mol. Biol. 1987, 193, 413–417. [Google Scholar] [CrossRef]
- Nackley, A.G.; Shabalina, S.A.; Tchivileva, I.E.; Satterfield, K.; Korchynskyi, O.; Makarov, S.S.; Maixner, W.; Diatchenko, L. Human catechol-o-methyltransferase haplotypes modulate protein expression by altering mrna secondary structure. Science 2006, 314, 1930–1933. [Google Scholar] [CrossRef]
- HLBI_Exome_Sequencing_Project. Exome Variant Server. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 9 January 2013).
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Chambers, J.C.; Zhao, J.; Terracciano, C.M.; Bezzina, C.R.; Zhang, W.; Kaba, R.; Navaratnarajah, M.; Lotlikar, A.; Sehmi, J.S.; Kooner, M.K.; et al. Genetic variation in scn10a influences cardiac conduction. Nat. Genet. 2010, 42, 149–152. [Google Scholar] [CrossRef]
- Pfeufer, A.; Sanna, S.; Arking, D.E.; Muller, M.; Gateva, V.; Fuchsberger, C.; Ehret, G.B.; Orru, M.; Pattaro, C.; Kottgen, A.; et al. Common variants at ten loci modulate the qt interval duration in the qtscd study. Nat. Genet. 2009, 41, 407–414. [Google Scholar] [CrossRef]
- Pfeufer, A.; Jalilzadeh, S.; Perz, S.; Mueller, J.C.; Hinterseer, M.; Illig, T.; Akyol, M.; Huth, C.; Schopfer-Wendels, A.; Kuch, B.; et al. Common variants in myocardial ion channel genes modify the qt interval in the general population: Results from the kora study. Circ. Res. 2005, 96, 693–701. [Google Scholar] [CrossRef]
- Ozawa, T.; Ito, M.; Tamaki, S.; Yao, T.; Ashihara, T.; Kita, Y.; Okamura, T.; Ueshima, H.; Horie, M. Gender and age effects on ventricular repolarization abnormality in japanese general carriers of a g643s common single nucleotide polymorphism for the kcnq1 gene. Circ. J. Offic. J. Jpn. Circ. Soc. 2006, 70, 645–650. [Google Scholar]
- Zeng, Z.Y.; Pu, J.L.; Tan, C.; Teng, S.Y.; Chen, J.H.; Su, S.Y.; Zhou, X.Y.; Zhang, S.; Li, Y.S.; Wang, F.Z.; et al. The association of single nucleotide polymorphism of slow delayed rectifier k+ channel genes with atrial fibrillation in han nationality chinese. Zhonghua Xin Xue Guan Bing Za Zhi 2005, 33, 987–991. [Google Scholar]
- Olszak-Waskiewicz, M.; Kubik, L.; Dziuk, M.; Sidlo, E.; Kucharczyk, K.; Kaczanowski, R. The association between scn5a, kcnq1 and kcne1 gene polymorphisms and complex ventricular arrhythmias in survivors of myocardial infarction. Kardiol. Pol. 2008, 66, 845–853. [Google Scholar]
- Tamargo, J.; Caballero, R.; Gomez, R.; Delpon, E. Cardiac electrophysiological effects of nitric oxide. Cardiovasc. Res. 2010, 87, 593–600. [Google Scholar] [CrossRef]
- Lima, B.; Forrester, M.T.; Hess, D.T.; Stamler, J.S. S-nitrosylation in cardiovascular signaling. Circ. Res. 2010, 106, 633–646. [Google Scholar] [CrossRef]
- Herron, T.J.; Devaney, E.J.; Metzger, J.M. Modulation of cardiac performance by motor protein gene transfer. Ann. N. Y. Acad. Sci. 2008, 1123, 96–104. [Google Scholar]
- Watkins, H.; Rosenzweig, A.; Hwang, D.S.; Levi, T.; McKenna, W.; Seidman, C.E.; Seidman, J.G. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N. Engl. J. Med. 1992, 326, 1108–1114. [Google Scholar] [CrossRef]
- Yu, Q.T.; Ifegwu, J.; Marian, A.J.; Mares, A., Jr.; Hill, R.; Perryman, M.B.; Bachinski, L.L.; Roberts, R. Hypertrophic cardiomyopathy mutation is expressed in messenger rna of skeletal as well as cardiac muscle. Circulation 1993, 87, 406–412. [Google Scholar] [CrossRef]
- Mi, H.; Lazareva-Ulitsky, B.; Loo, R.; Kejariwal, A.; Vandergriff, J.; Rabkin, S.; Guo, N.; Muruganujan, A.; Doremieux, O.; Campbell, M.J.; et al. The panther database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005, 33, D284–D288. [Google Scholar] [CrossRef]
- You, J.J.; Singer, D.E.; Howard, P.A.; Lane, D.A.; Eckman, M.H.; Fang, M.C.; Hylek, E.M.; Schulman, S.; Go, A.S.; Hughes, M.; et al. Antithrombotic therapy for atrial fibrillation: Antithrombotic therapy and prevention of thrombosis, 9th ed: American college of chest physicians evidence-based clinical practice guidelines. Chest 2012, 141, e531S–e575S. [Google Scholar]
- Hein, S.; Kostin, S.; Heling, A.; Maeno, Y.; Schaper, J. The role of the cytoskeleton in heart failure. Cardiovasc. Res. 2000, 45, 273–278. [Google Scholar] [CrossRef]
- Roberts, R.; Schwartz, K. Myocardial diseases. Circulation 2000, 102, IV34–IV39. [Google Scholar]
- Heling, A.; Zimmermann, R.; Kostin, S.; Maeno, Y.; Hein, S.; Devaux, B.; Bauer, E.; Klovekorn, W.P.; Schlepper, M.; Schaper, W.; et al. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ. Res. 2000, 86, 846–853. [Google Scholar] [CrossRef]
- Xiao, J.; Cao, H.; Liang, D.; Liu, Y.; Zhang, H.; Zhao, H.; Li, J.; Yan, B.; Peng, L.; Zhou, Z.; et al. Taxol, a microtubule stabilizer, prevents ischemic ventricular arrhythmias in rats. J. Cell. Mol. Med. 2011, 15, 1166–1176. [Google Scholar] [CrossRef]
- Simonyan, V.; Mazumder, R. High-performance integrated virtual environment clouds (hive) for extra-large (xl) data analysis. Comparative sequence, genome analysis, genome assembly, & genome scale computational methods session. In Proceesings of the 2011 International Conference on Bioinformatics and Computational Biology, Las Vegas, NV, USA, 17–21 July 2011.
- Wu, T.-J.; Shamsaddini, A.; Pan, Y.; Smith, K.; Crichton, D.; Simonyan, V.; Mazumder, R. A framework for organizing cancer related variations from existing databases, publications and ngs data using a high-performance integrated virtual environment (HIVE). Database 2014, in press. [Google Scholar]
- Johnston, J.J.; Rubinstein, W.S.; Facio, F.M.; Ng, D.; Singh, L.N.; Teer, J.K.; Mullikin, J.C.; Biesecker, L.G. Secondary variants in individuals undergoing exome sequencing: Screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibili ty genes. Am. J. Hum. Genet. 2012, 91, 97–108. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abunimer, A.; Smith, K.; Wu, T.-J.; Lam, P.; Simonyan, V.; Mazumder, R. Single-Nucleotide Variations in Cardiac Arrhythmias: Prospects for Genomics and Proteomics Based Biomarker Discovery and Diagnostics. Genes 2014, 5, 254-269. https://doi.org/10.3390/genes5020254
Abunimer A, Smith K, Wu T-J, Lam P, Simonyan V, Mazumder R. Single-Nucleotide Variations in Cardiac Arrhythmias: Prospects for Genomics and Proteomics Based Biomarker Discovery and Diagnostics. Genes. 2014; 5(2):254-269. https://doi.org/10.3390/genes5020254
Chicago/Turabian StyleAbunimer, Ayman, Krista Smith, Tsung-Jung Wu, Phuc Lam, Vahan Simonyan, and Raja Mazumder. 2014. "Single-Nucleotide Variations in Cardiac Arrhythmias: Prospects for Genomics and Proteomics Based Biomarker Discovery and Diagnostics" Genes 5, no. 2: 254-269. https://doi.org/10.3390/genes5020254
APA StyleAbunimer, A., Smith, K., Wu, T.-J., Lam, P., Simonyan, V., & Mazumder, R. (2014). Single-Nucleotide Variations in Cardiac Arrhythmias: Prospects for Genomics and Proteomics Based Biomarker Discovery and Diagnostics. Genes, 5(2), 254-269. https://doi.org/10.3390/genes5020254