In Vivo RNAi-Based Screens: Studies in Model Organisms

Abstract
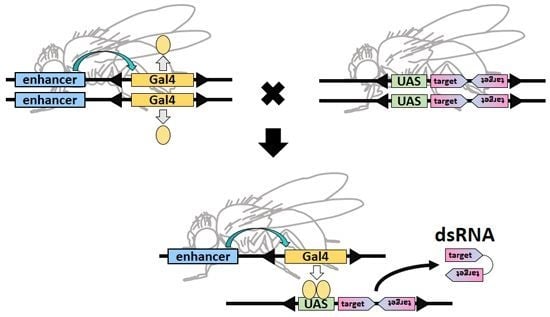
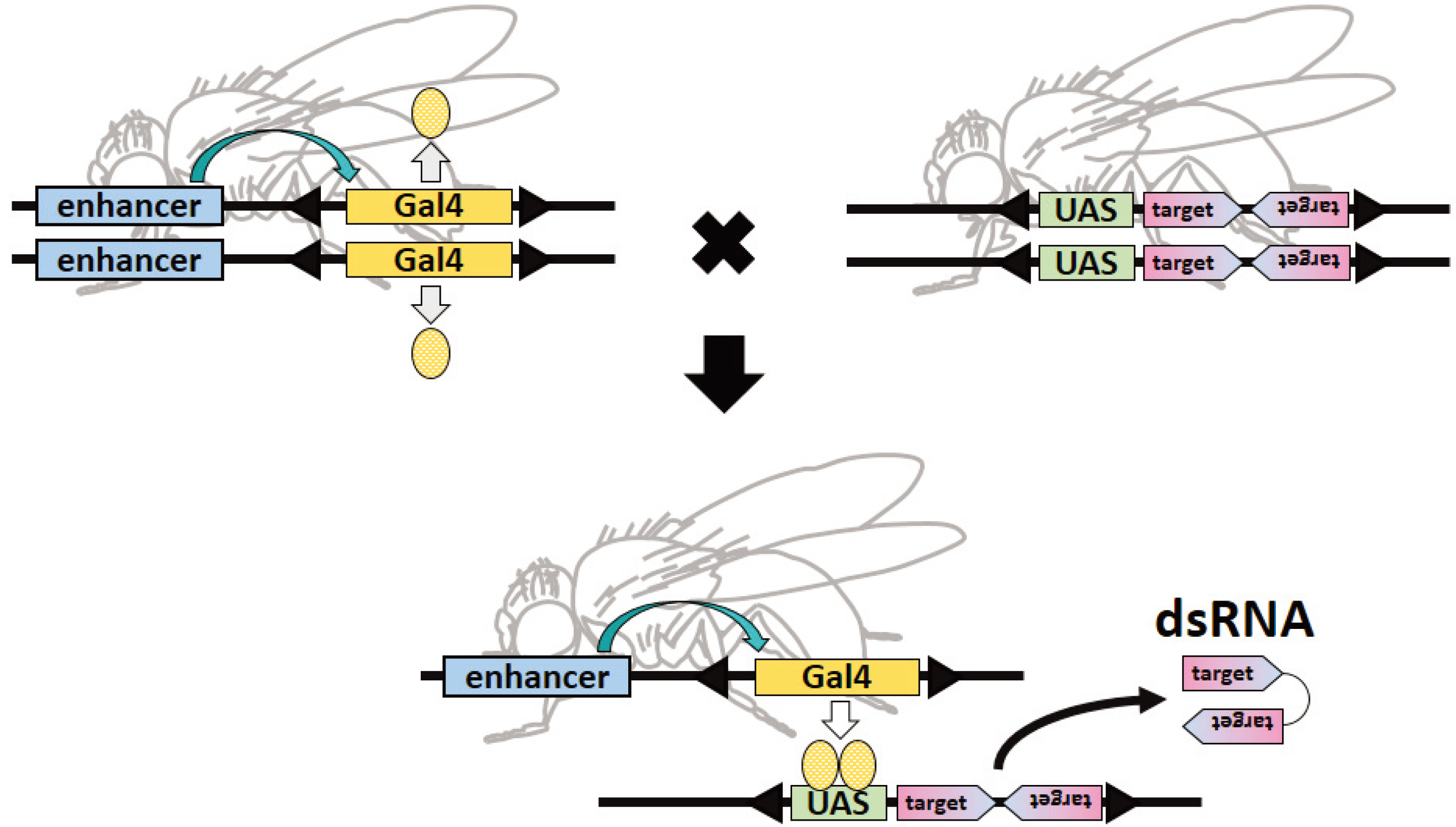
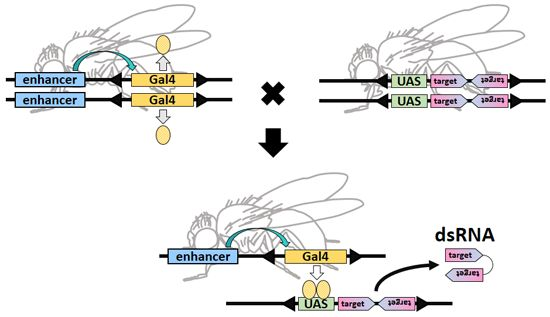
:
1. Introduction
2. RNA Interference
3. Delivery and/or Expression of dsRNAs in Target Cells
4. Genome-Wide RNAi in Drosophila Cultured Cells
5. Drosophila RNAi Libraries
5.1. Advantages

{kind=link}
{kind=link}
5.2. Disadvantages
6. Genome-Wide In Vivo RNAi in Drosophila
| Class | Authors | Purpose: What kinds of genes are expected to be identified | Reference |
|---|---|---|---|
| Class 1 | Cronin et al. | genes against intestinal infection with Serratia marcescens | [56] |
| Osman et al. | suppressors of AML1-ETO | [57] | |
| Yamamoto-Hino et al. | genes involved in gylcosylation | [58] | |
| Avet-Rochex et al | melanotic tumor suppressor genes involved in blood cell homeostasis | [59] | |
| Neely et al. | genes involved in heart development and function | [60] | |
| Neely et al. | genes regulating pain | [61] | |
| Lesch et al. | genes required for wound closure | [62] | |
| Schnorrer et al. | genes involved in muscle morphogenesis and function | [63] | |
| Pospisilik et al. | genes involved in obesity | [64] | |
| Neumuller et al. | genes involved in stem cell differentiation | [65] | |
| Yano et al. | genes involved in apical transport | [66] | |
| Carney et al. | genes maintaining proper neuroblast numbers | [67] | |
| Valakh et al. | genes involved in formation, growth, and maintenance of the neuromuscular junction | [68] | |
| Class 2 | Mummery-Widmer et al. | Notch regulators | [69] |
| Vosfeldt et al. | modifiers of polyQ dependent toxicity | [70] | |
| Llamusi et al. | modifiers of CTG repeat dependent toxicity | [71] | |
| Class 3 | Kambris et al. | serine protease genes required for Toll activation | [72] |
| Saj et al. | Notch regulators | [73] | |
| Port et al. | genes involved in Wg secretion | [74] | |
| Du et al. | regulators of Hh pathway | [75] | |
| Aikin et al. | genes involved in Hh secretion | [76] |
6.1. Class 1: Screens to Address Questions that Can only Be Investigated Using an In Vivo Approach
6.2. Class 2: Screens to Identify New Genes that Can Be Found by both In Vivo and In Vitro Experiments
6.3. Class 3: Screens to Find True Positives among Candidate Genes Identified in In Vitro Screens
7. Erroneous Results and Possible Solutions
8. In Vivo RNAi in Mice
| Authors | Purpose: what kinds of genes are expected to be identified | Number of shRNAs | Reference |
|---|---|---|---|
| Zender et al. | genes involved in hepatocarcinogenesis | 631 | [96] |
| Bric et al. | genes involved in lymphomagenesis | 2,300 | [97] |
| Maecham et al. | genes involved in lymphoma prgression | 2,250 | [98] |
| Wuestefeld et al. | genes involved in liver regeneration | 631 | [99] |
| Vaeble et al. | genes involved in viral replication | >10,000 | [100] |
| Beronja et al. | genes involved in epidermal growth | >77,000 | [101] |
9. Conclusions
Acknowledgments
Conflicts of interest
References
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 1998, 282, 2012–2018. [Google Scholar] [CrossRef]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The genome sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef]
- Silva, J.; Chang, K.; Hannon, G.J.; Rivas, F.V. RNA-interference-based functional genomics in mammalian cells: Reverse genetics coming of age. Oncogene 2004, 23, 8401–8409. [Google Scholar] [CrossRef]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef]
- Whitehurst, A.W.; Bodemann, B.O.; Cardenas, J.; Ferguson, D.; Girard, L.; Peyton, M.; Minna, J.D.; Michnoff, C.; Hao, W.; Roth, M.G.; et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007, 446, 815–819. [Google Scholar] [CrossRef]
- Boutros, M.; Kiger, A.A.; Armknecht, S.; Kerr, K.; Hild, M.; Koch, B.; Haas, S.A.; Paro, R.; Perrimon, N. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 2004, 303, 832–835. [Google Scholar] [CrossRef]
- Ulvila, J.; Parikka, M.; Kleino, A.; Sormunen, R.; Ezekowitz, R.A.; Kocks, C.; Ramet, M. Double-stranded RNA is internalized by scavenger receptor-mediated endocytosis in Drosophila S2 cells. J. Biol. Chem. 2006, 281, 14370–14375. [Google Scholar]
- Timmons, L.; Fire, A. Specific interference by ingested dsRNA. Nature 1998, 395, 854. [Google Scholar] [CrossRef]
- Tabara, H.; Grishok, A.; Mello, C.C. RNAi in C. elegans: Soaking in the genome sequence. Science 1998, 282, 430–431. [Google Scholar] [CrossRef]
- Maeda, I.; Kohara, Y.; Yamamoto, M.; Sugimoto, A. Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr. Biol. 2001, 11, 171–176. [Google Scholar] [CrossRef]
- Lehner, B.; Tischler, J.; Fraser, A.G. RNAi screens in Caenorhabditis elegans in a 96-well liquid format and their application to the systematic identification of genetic interactions. Nat. Protoc. 2006, 1, 1617–1620. [Google Scholar] [CrossRef]
- Kennerdell, J.R.; Carthew, R.W. Use of dsRNA-mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell 1998, 95, 1017–1026. [Google Scholar] [CrossRef]
- Misquitta, L.; Paterson, B.M. Targeted disruption of gene function in Drosophila by RNA interference (RNA-i): A role for nautilus in embryonic somatic muscle formation. Proc. Natl. Acad. Sci. USA 1999, 96, 1451–1456. [Google Scholar] [CrossRef]
- Lam, G.; Thummel, C.S. Inducible expression of double-stranded RNA directs specific genetic interference in Drosophila. Curr. Biol. 2000, 10, 957–963. [Google Scholar] [CrossRef]
- Fortier, E.; Belote, J.M. Temperature-dependent gene silencing by an expressed inverted repeat in Drosophila. Genesis 2000, 26, 240–244. [Google Scholar] [CrossRef]
- Martinek, S.; Young, M.W. Specific genetic interference with behavioral rhythms in Drosophila by expression of inverted repeats. Genetics 2000, 156, 1717–1725. [Google Scholar]
- Giordano, E.; Rendina, R.; Peluso, I.; Furia, M. RNAi triggered by symmetrically transcribed transgenes in Drosophila melanogaster. Genetics 2002, 160, 637–648. [Google Scholar]
- Dietzl, G.; Chen, D.; Schnorrer, F.; Su, K.C.; Barinova, Y.; Fellner, M.; Gasser, B.; Kinsey, K.; Oppel, S.; Scheiblauer, S.; et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007, 448, 151–156. [Google Scholar] [CrossRef]
- NIG-FLY. Available online: http://www.shigen.nig.ac.jp/fly/nigfly/about/aboutRnai.jsp/ (accessed on 19 November 2013).
- Transgenic RNAi Project. Available online: http://www.flyrnai.org/TRiP-HOME.html/ (accessed on 19 November 2013).
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef]
- Karpala, A.J.; Doran, T.J.; Bean, A.G. Immune responses to dsRNA: Implications for gene silencing technologies. Immunol. Cell Biol. 2005, 83, 211–216. [Google Scholar] [CrossRef]
- Gantier, M.P.; Williams, B.R. The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev. 2007, 18, 363–371. [Google Scholar] [CrossRef]
- Bantounas, I.; Phylactou, L.A.; Uney, J.B. RNA interference and the use of small interfering RNA to study gene function in mammalian systems. J. Mol. Endocrinol. 2004, 33, 545–557. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Yates, L.A.; Norbury, C.J.; Gilbert, R.J. The long and short of microRNA. Cell 2013, 153, 516–519. [Google Scholar] [CrossRef]
- Chang, K.; Elledge, S.J.; Hannon, G.J. Lessons from Nature: MicroRNA-based shRNA libraries. Nat. Methods 2006, 3, 707–714. [Google Scholar] [CrossRef]
- Ni, J.Q.; Zhou, R.; Czech, B.; Liu, L.P.; Holderbaum, L.; Yang-Zhou, D.; Shim, H.S.; Tao, R.; Handler, D.; Karpowicz, P.; et al. A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat. Methods 2011, 8, 405–407. [Google Scholar] [CrossRef]
- Sandy, P.; Ventura, A.; Jacks, T. Mammalian RNAi: A practical guide. Biotechniques 2005, 39, 215–224. [Google Scholar] [CrossRef]
- Couto, L.B.; High, K.A. Viral vector-mediated RNA interference. Curr. Opin. Pharmacol. 2010, 10, 534–542. [Google Scholar] [CrossRef]
- Pan, Q.; van der Laan, L.J.; Janssen, H.L.; Peppelenbosch, M.P. A dynamic perspective of RNAi library development. Trends Biotechnol. 2012, 30, 206–215. [Google Scholar] [CrossRef]
- Mohr, S.; Bakal, C.; Perrimon, N. Genomic screening with RNAi: Results and challenges. Annu. Rev. Biochem. 2010, 79, 37–64. [Google Scholar] [CrossRef]
- Flockhart, I.T.; Booker, M.; Hu, Y.; McElvany, B.; Gilly, Q.; Mathey-Prevot, B.; Perrimon, N.; Mohr, S.E. FlyRNAi.org—The database of the Drosophila RNAi screening center: 2012 update. Nucleic Acids Res. 2012, 40, D715–D719. [Google Scholar] [CrossRef]
- Schmidt, E.E.; Pelz, O.; Buhlmann, S.; Kerr, G.; Horn, T.; Boutros, M. GenomeRNAi: A database for cell-based and in vivo RNAi phenotypes, 2013 update. Nucleic Acids Res. 2013, 41, D1021–D1026. [Google Scholar]
- Liu, T.; Sims, D.; Baum, B. Parallel RNAi screens across different cell lines identify generic and cell type-specific regulators of actin organization and cell morphology. Genome Biol. 2009, 10, R26. [Google Scholar] [CrossRef]
- Sepp, K.J.; Hong, P.; Lizarraga, S.B.; Liu, J.S.; Mejia, L.A.; Walsh, C.A.; Perrimon, N. Identification of neural outgrowth genes using genome-wide RNAi. PLoS Genet. 2008, 4, e1000111. [Google Scholar] [CrossRef]
- Hayashi, S.; Ito, K.; Sado, Y.; Taniguchi, M.; Akimoto, A.; Takeuchi, H.; Aigaki, T.; Matsuzaki, F.; Nakagoshi, H.; Tanimura, T.; et al. GETDB, a database compiling expression patterns and molecular locations of a collection of Gal4 enhancer traps. Genesis 2002, 34, 58–61. [Google Scholar] [CrossRef]
- Drosophila Genetic Resource Center. Available online: http://www.dgrc.kit.ac.jp/ (accessed on 19 November 2013).
- Yamamoto-Hino, M.; Goto, S. Rikkyo University: Tokyo, Japan, 2013; Unpublished work.
- Vinna Drosophila RNAi Center. Available online: http://stockcenter.vdrc.at/control/main/ (accessed on 19 November 2013).
- dsCheck. Available online: http://dscheck.rnai.jp/ (accessed on 19 November 2013).
- Vinna Drosophila RNAi Center. Definition. Available online: http://stockcenter.vdrc.at/control/vdrcdefinition/ (accessed on 19 November 2013).
- Meadows, L. Personal communication, Vinna Drosophila RNAi center: Vienna, Austria, 2013.
- Research Resource Circulation. Available online: http://rrc.nbrp.jp/index.jsp/ (accessed on 19 November 2013).
- Cronin, S.J.; Nehme, N.T.; Limmer, S.; Liegeois, S.; Pospisilik, J.A.; Schramek, D.; Leibbrandt, A.; Simoes Rde, M.; Gruber, S.; Puc, U.; et al. Genome-wide RNAi screen identifies genes involved in intestinal pathogenic bacterial infection. Science 2009, 325, 340–343. [Google Scholar] [CrossRef]
- Osman, D.; Gobert, V.; Ponthan, F.; Heidenreich, O.; Haenlin, M.; Waltzer, L. A Drosophila model identifies calpains as modulators of the human leukemogenic fusion protein AML1-ETO. Proc. Natl. Acad. Sci. USA 2009, 106, 12043–12048. [Google Scholar]
- Yamamoto-Hino, M.; Kanie, Y.; Awano, W.; Aoki-Kinoshita, K.F.; Yano, H.; Nishihara, S.; Okano, H.; Ueda, R.; Kanie, O.; Goto, S. Identification of genes required for neural-specific glycosylation using functional genomics. PLoS Genet. 2010, 6, e1001254. [Google Scholar]
- Avet-Rochex, A.; Boyer, K.; Polesello, C.; Gobert, V.; Osman, D.; Roch, F.; Auge, B.; Zanet, J.; Haenlin, M.; Waltzer, L. An in vivo RNA interference screen identifies gene networks controlling Drosophila melanogaster blood cell homeostasis. BMC Dev. Biol. 2010, 10, 65. [Google Scholar] [CrossRef]
- Neely, G.G.; Kuba, K.; Cammarato, A.; Isobe, K.; Amann, S.; Zhang, L.; Murata, M.; Elmen, L.; Gupta, V.; Arora, S.; et al. A global in vivo Drosophila RNAi screen identifies NOT3 as a conserved regulator of heart function. Cell 2010, 141, 142–153. [Google Scholar] [CrossRef]
- Neely, G.G.; Hess, A.; Costigan, M.; Keene, A.C.; Goulas, S.; Langeslag, M.; Griffin, R.S.; Belfer, I.; Dai, F.; Smith, S.B.; et al. A genome-wide Drosophila screen for heat nociception identifies alpha2delta3 as an evolutionarily conserved pain gene. Cell 2010, 143, 628–638. [Google Scholar] [CrossRef]
- Lesch, C.; Jo, J.; Wu, Y.; Fish, G.S.; Galko, M.J. A targeted UAS-RNAi screen in Drosophila larvae identifies wound closure genes regulating distinct cellular processes. Genetics 2010, 186, 943–957. [Google Scholar] [CrossRef]
- Schnorrer, F.; Schonbauer, C.; Langer, C.C.; Dietzl, G.; Novatchkova, M.; Schernhuber, K.; Fellner, M.; Azaryan, A.; Radolf, M.; Stark, A.; et al. Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature 2010, 464, 287–291. [Google Scholar]
- Pospisilik, J.A.; Schramek, D.; Schnidar, H.; Cronin, S.J.; Nehme, N.T.; Zhang, X.; Knauf, C.; Cani, P.D.; Aumayr, K.; Todoric, J.; et al. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell 2010, 140, 148–160. [Google Scholar] [CrossRef]
- Neumuller, R.A.; Richter, C.; Fischer, A.; Novatchkova, M.; Neumuller, K.G.; Knoblich, J.A. Genome-wide analysis of self-renewal in Drosophila neural stem cells by transgenic RNAi. Cell Stem Cell 2011, 8, 580–593. [Google Scholar] [CrossRef]
- Yano, H.; Yamamoto-Hino, M.; Awano, W.; Aoki-Kinoshita, K.F.; Tsuda-Sakurai, K.; Okano, H.; Goto, S. Identification of proteasome components required for apical localization of Chaoptin using functional genomics. J. Neurogenet. 2012, 26, 53–63. [Google Scholar] [CrossRef]
- Carney, T.D.; Miller, M.R.; Robinson, K.J.; Bayraktar, O.A.; Osterhout, J.A.; Doe, C.Q. Functional genomics identifies neural stem cell sub-type expression profiles and genes regulating neuroblast homeostasis. Dev. Biol. 2012, 361, 137–146. [Google Scholar] [CrossRef]
- Valakh, V.; Naylor, S.A.; Berns, D.S.; DiAntonio, A. A large-scale RNAi screen identifies functional classes of genes shaping synaptic development and maintenance. Dev. Biol. 2012, 366, 163–171. [Google Scholar] [CrossRef]
- Mummery-Widmer, J.L.; Yamazaki, M.; Stoeger, T.; Novatchkova, M.; Bhalerao, S.; Chen, D.; Dietzl, G.; Dickson, B.J.; Knoblich, J.A. Genome-wide analysis of Notch signalling in Drosophila by transgenic RNAi. Nature 2009, 458, 987–992. [Google Scholar] [CrossRef]
- Vo, S.H.; Butzlaff, M.; Pru, S.K.; Ni Charthaigh, R.A.; Karsten, P.; Lankes, A.; Hamm, S.; Simons, M.; Adryan, B.; Schulz, J.B.; et al. Large-scale screen for modifiers of ataxin-3-derived polyglutamine-induced toxicity in Drosophila. PLoS One 2012, 7, e47452. [Google Scholar] [CrossRef]
- Llamusi, B.; Bargiela, A.; Fernandez-Costa, J.M.; Garcia-Lopez, A.; Klima, R.; Feiguin, F.; Artero, R. Muscleblind, BSF and TBPH are mislocalized in the muscle sarcomere of a Drosophila myotonic dystrophy model. Dis. Model. Mech. 2013, 6, 184–196. [Google Scholar]
- Kambris, Z.; Brun, S.; Jang, I.H.; Nam, H.J.; Romeo, Y.; Takahashi, K.; Lee, W.J.; Ueda, R.; Lemaitre, B. Drosophila immunity: A large-scale in vivo RNAi screen identifies five serine proteases required for Toll activation. Curr. Biol. 2006, 16, 808–813. [Google Scholar] [CrossRef]
- Saj, A.; Arziman, Z.; Stempfle, D.; van Belle, W.; Sauder, U.; Horn, T.; Durrenberger, M.; Paro, R.; Boutros, M.; Merdes, G. A combined ex vivo and in vivo RNAi screen for notch regulators in Drosophila reveals an extensive notch interaction network. Dev. Cell. 2010, 18, 862–876. [Google Scholar] [CrossRef]
- Port, F.; Hausmann, G.; Basler, K. A genome-wide RNA interference screen uncovers two p24 proteins as regulators of Wingless secretion. EMBO Rep. 2011, 12, 1144–1152. [Google Scholar] [CrossRef]
- Du, J.; Zhang, J.; Su, Y.; Liu, M.; Ospina, J.K.; Yang, S.; Zhu, A.J. In vivo RNAi screen reveals neddylation genes as novel regulators of Hedgehog signaling. PLoS One 2011, 6, e24168. [Google Scholar]
- Aikin, R.; Cervantes, A.; D'Angelo, G.; Ruel, L.; Lacas-Gervais, S.; Schaub, S.; Therond, P. A genome-wide RNAi screen identifies regulators of cholesterol-modified hedgehog secretion in Drosophila. PLoS One 2012, 7, e33665. [Google Scholar]
- Costello, J.C.; Dalkilic, M.M.; Beason, S.M.; Gehlhausen, J.R.; Patwardhan, R.; Middha, S.; Eads, B.D.; Andrews, J.R. Gene networks in Drosophila melanogaster: Integrating experimental data to predict gene function. Genome Biol. 2009, 10, R97. [Google Scholar] [CrossRef]
- Bilen, J.; Bonini, N.M. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 2007, 3, 1950–1964. [Google Scholar]
- Zhang, S.; Binari, R.; Zhou, R.; Perrimon, N. A genomewide RNA interference screen for modifiers of aggregates formation by mutant Huntingtin in Drosophila. Genetics 2010, 184, 1165–1179. [Google Scholar] [CrossRef]
- Nollen, E.A.; Garcia, S.M.; van Haaften, G.; Kim, S.; Chavez, A.; Morimoto, R.I.; Plasterk, R.H. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA 2004, 101, 6403–6408. [Google Scholar]
- Jang, I.H.; Chosa, N.; Kim, S.H.; Nam, H.J.; Lemaitre, B.; Ochiai, M.; Kambris, Z.; Brun, S.; Hashimoto, C.; Ashida, M.; et al. A Spatzle-processing enzyme required for toll signaling activation in Drosophila innate immunity. Dev. Cell 2006, 10, 45–55. [Google Scholar]
- Horn, T.; Arziman, Z.; Berger, J.; Boutros, M. GenomeRNAi: A database for cell-based RNAi phenotypes. Nucleic Acids Res. 2007, 35, D492–D497. [Google Scholar] [CrossRef]
- Flybase. Available online: http://flybase.org/ (accessed on 19 November 2013).
- Echeverri, C.J.; Beachy, P.A.; Baum, B.; Boutros, M.; Buchholz, F.; Chanda, S.K.; Downward, J.; Ellenberg, J.; Fraser, A.G.; Hacohen, N.; et al. Minimizing the risk of reporting false positives in large-scale RNAi screens. Nat. Methods 2006, 3, 777–779. [Google Scholar] [CrossRef]
- Kondo, S.; Booker, M.; Perrimon, N. Cross-species RNAi rescue platform in Drosophila melanogaster. Genetics 2009, 183, 1165–1173. [Google Scholar] [CrossRef]
- Booker, M.; Samsonova, A.A.; Kwon, Y.; Flockhart, I.; Mohr, S.E.; Perrimon, N. False negative rates in Drosophila cell-based RNAi screens: A case study. BMC Genomics 2011, 12, 50. [Google Scholar] [CrossRef]
- Groth, A.C.; Fish, M.; Nusse, R.; Calos, M.P. Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics 2004, 166, 1775–1782. [Google Scholar] [CrossRef]
- Bateman, J.R.; Lee, A.M.; Wu, C.T. Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics 2006, 173, 769–777. [Google Scholar] [CrossRef]
- Venken, K.J.; He, Y.; Hoskins, R.A.; Bellen, H.J. P[acman]: A BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 2006, 314, 1747–1751. [Google Scholar] [CrossRef]
- Bischof, J.; Maeda, R.K.; Hediger, M.; Karch, F.; Basler, K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. USA 2007, 104, 3312–3317. [Google Scholar] [CrossRef]
- Markstein, M.; Pitsouli, C.; Villalta, C.; Celniker, S.E.; Perrimon, N. Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat. Genet. 2008, 40, 476–483. [Google Scholar] [CrossRef]
- Pfeiffer, B.D.; Ngo, T.T.; Hibbard, K.L.; Murphy, C.; Jenett, A.; Truman, J.W.; Rubin, G.M. Refinement of tools for targeted gene expression in Drosophila. Genetics 2010, 186, 735–755. [Google Scholar] [CrossRef]
- Kondo, S.; Ueda, R. Personal communication, National institute of genetics: Mishima, Japan, 2013.
- Wang, L.; Tu, Z.; Sun, F. A network-based integrative approach to prioritize reliable hits from multiple genome-wide RNAi screens in Drosophila. BMC Genomics 2009, 10, 220. [Google Scholar] [CrossRef]
- Guest, S.T.; Yu, J.; Liu, D.; Hines, J.A.; Kashat, M.A.; Finley, R.L., Jr. A protein network-guided screen for cell cycle regulators in Drosophila. BMC Syst. Biol. 2011, 5, 65. [Google Scholar] [CrossRef]
- Zender, L.; Xue, W.; Zuber, J.; Semighini, C.P.; Krasnitz, A.; Ma, B.; Zender, P.; Kubicka, S.; Luk, J.M.; Schirmacher, P.; et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 2008, 135, 852–864. [Google Scholar]
- Bric, A.; Miething, C.; Bialucha, C.U.; Scuoppo, C.; Zender, L.; Krasnitz, A.; Xuan, Z.; Zuber, J.; Wigler, M.; Hicks, J.; et al. Functional identification of tumor-suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell 2009, 16, 324–335. [Google Scholar] [CrossRef]
- Meacham, C.E.; Ho, E.E.; Dubrovsky, E.; Gertler, F.B.; Hemann, M.T. In vivo RNAi screening identifies regulators of actin dynamics as key determinants of lymphoma progression. Nat. Genet. 2009, 41, 1133–1137. [Google Scholar] [CrossRef]
- Wuestefeld, T.; Pesic, M.; Rudalska, R.; Dauch, D.; Longerich, T.; Kang, T.W.; Yevsa, T.; Heinzmann, F.; Hoenicke, L.; Hohmeyer, A.; et al. A Direct in vivo RNAi screen identifies MKK4 as a key regulator of liver regeneration. Cell 2013, 153, 389–401. [Google Scholar] [CrossRef]
- Varble, A.; Benitez, A.A.; Schmid, S.; Sachs, D.; Shim, J.V.; Rodriguez-Barrueco, R.; Panis, M.; Crumiller, M.; Silva, J.M.; Sachidanandam, R.; et al. An in vivo RNAi screening approach to identify host determinants of virus replication. Cell Host Microbe 2013, 14, 346–356. [Google Scholar]
- Beronja, S.; Janki, P.; Heller, E.; Lien, W.H.; Keyes, B.E.; Oshimori, N.; Fuchs, E. RNAi screens in mice identify physiological regulators of oncogenic growth. Nature 2013, 501, 185–190. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yamamoto-Hino, M.; Goto, S. In Vivo RNAi-Based Screens: Studies in Model Organisms. Genes 2013, 4, 646-665. https://doi.org/10.3390/genes4040646
Yamamoto-Hino M, Goto S. In Vivo RNAi-Based Screens: Studies in Model Organisms. Genes. 2013; 4(4):646-665. https://doi.org/10.3390/genes4040646
Chicago/Turabian StyleYamamoto-Hino, Miki, and Satoshi Goto. 2013. "In Vivo RNAi-Based Screens: Studies in Model Organisms" Genes 4, no. 4: 646-665. https://doi.org/10.3390/genes4040646
APA StyleYamamoto-Hino, M., & Goto, S. (2013). In Vivo RNAi-Based Screens: Studies in Model Organisms. Genes, 4(4), 646-665. https://doi.org/10.3390/genes4040646