The Human Transcriptome: An Unfinished Story

Abstract
:1. Background and Introduction
2. The Diversity of the Transcriptome
2.1. Various Classes of ncRNAs
- - they are expressed in a highly tissue-specific manner compared to protein-coding genes,
- - they are typically co-expressed with their neighboring genes, and
- - they only show moderate conservation in other species.
2.2. Alternative Splicing
2.3. Estimating the Annotated Human Transcript Count
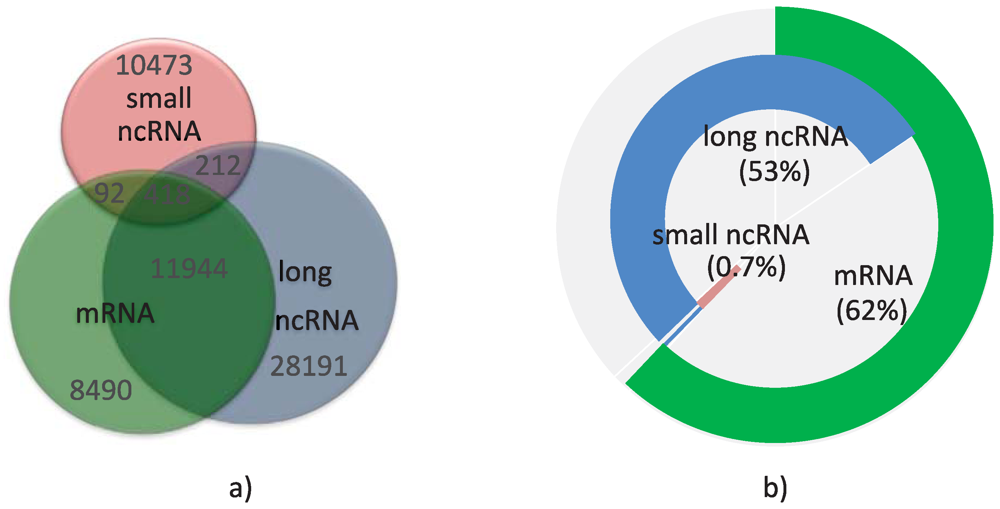

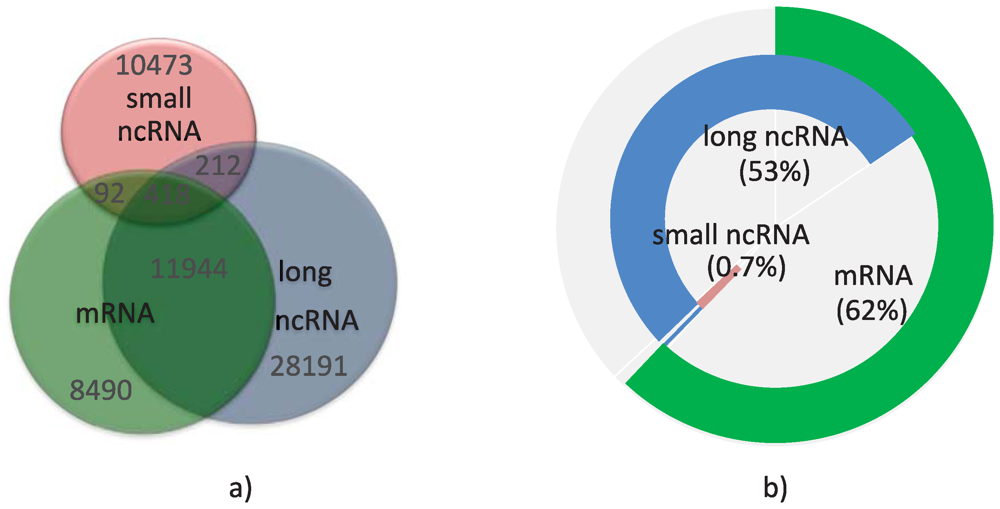{kind=link}
{kind=link}
| Annotation | mRNA | Long ncRNA | Small ncRNA |
|---|---|---|---|
| Transcripts | 111,451 | 89,981 | 11,366 |
| Loci | 20,944 | 40,765 | 11,195 |
2.4. RNA Editing
3. Reconstructing the Transcriptome
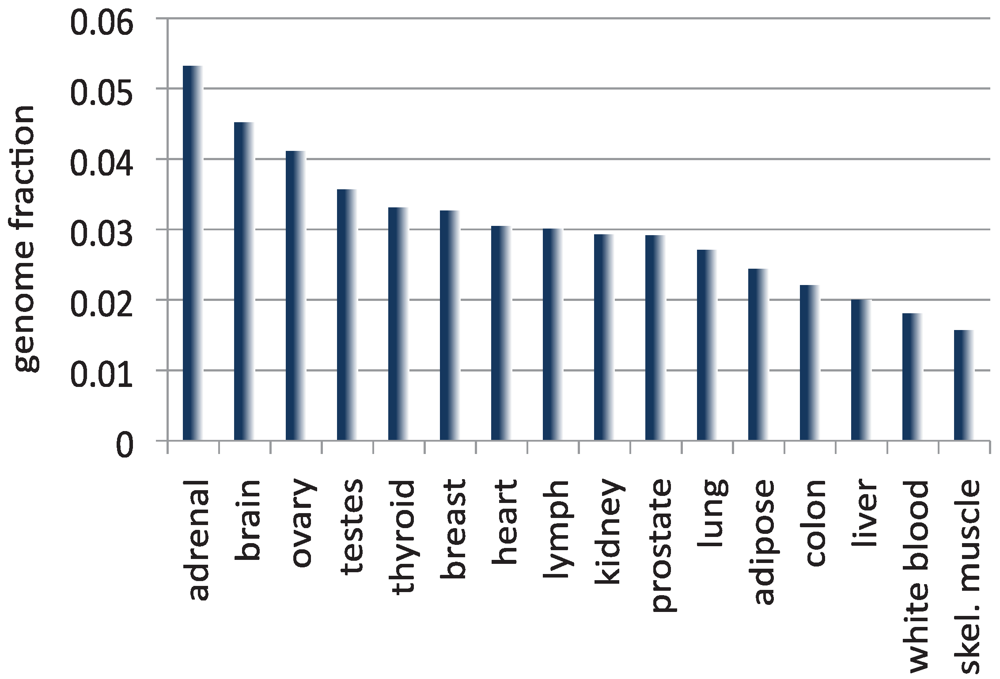
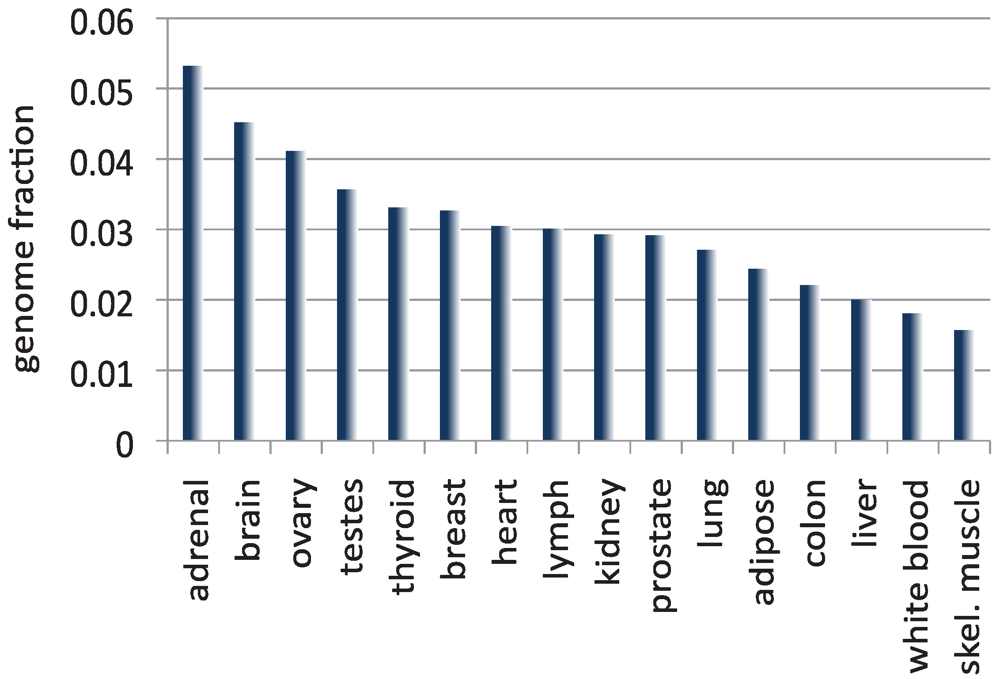
4. The Size of the Transcriptome
5. Discussion and Conclusions
Acknowledgements
References
- Ohno, S. So much “junk” DNA in our genome. Brookhaven Symp. Biol. 1972, 23, 366–370. [Google Scholar]
- The International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar]
- Chen, J.; Sun, M.; Lee, S.; Zhou, G.; Rowley, J.D.; Wang, S.M. Identifying novel transcripts and novel genes in the human genome by using novel SAGE tags. Proc. Natl. Acad. Sci. USA 2002, 99, 12257–12262. [Google Scholar]
- Kapranov, P.; Cawley, S.E.; Drenkow, J.; Bekiranov, S.; Strausberg, R.L.; Fodor, S.P.; Gingeras, T.R. Large-scale transcriptional activity in chromosomes 21 and 22. Science 2002, 296, 916–919. [Google Scholar] [CrossRef]
- Saha, S.; Sparks, A.B.; Rago, C.; Akmaev, V.; Wang, C.J.; Vogelstein, B.; Kinzler, K.W.; Velculescu, V.E. Using the transcriptome to annotate the genome. Nat. Biotechnol. 2002, 20, 508–512. [Google Scholar] [CrossRef]
- Mattick, J.S. The central role of RNA in human development and cognition. FEBS Lett. 2011, 585, 1600–1616. [Google Scholar] [CrossRef]
- Griffin, H.G.; Griffin, A.M. DNA sequencing. Recent innovations and future trends. Appl. Biochem. Biotechnol. 1993, 38, 147–159. [Google Scholar] [CrossRef]
- Adams, M.D.; Kerlavage, A.R.; Fields, C.; Venter, J.C. 3,400 new expressed sequence tags identify diversity of transcripts in human brain. Nat. Genet. 1993, 4, 256–267. [Google Scholar] [CrossRef]
- Adams, M.D.; Kerlavage, A.R.; Fleischmann, R.D.; Fuldner, R.A.; Bult, C.J.; Lee, N.H.; Kirkness, E.F.; Weinstock, K.G.; Gocayne, J.D.; White, O.; et al. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature 1995, 377, 3–174. [Google Scholar]
- Pertea, M.; Salzberg, S.L. Between a chicken and a grape: Estimating the number of human genes. Genome Biol. 2010, 11, 206. [Google Scholar] [CrossRef]
- Strausberg, R.L.; Riggins, G.J. Navigating the human transcriptome. Proc. Natl. Acad. Sci. USA 2001, 98, 11837–11838. [Google Scholar] [CrossRef]
- Velculescu, V.E.; Zhang, L.; Vogelstein, B.; Kinzler, K.W. Serial analysis of gene expression. Science 1995, 270, 484–487. [Google Scholar]
- Shiraki, T.; Kondo, S.; Katayama, S.; Waki, K.; Kasukawa, T.; Kawaji, H.; Kodzius, R.; Watahiki, A.; Nakamura, M.; Arakawa, T.; et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. USA 2003, 100, 15776–15781. [Google Scholar]
- Brenner, S.; Johnson, M.; Bridgham, J.; Golda, G.; Lloyd, D.H.; Johnson, D.; Luo, S.; McCurdy, S.; Foy, M.; Ewan, M.; et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat. Biotechnol. 2000, 18, 630–634. [Google Scholar]
- Clark, T.A.; Sugnet, C.W.; Ares, M., Jr. Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science 2002, 296, 907–910. [Google Scholar]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar]
- Lashkari, D.A.; DeRisi, J.L.; McCusker, J.H.; Namath, A.F.; Gentile, C.; Hwang, S.Y.; Brown, P.O.; Davis, R.W. Yeast microarrays for genome wide parallel genetic and gene expression analysis. Proc. Natl. Acad. Sci. USA 1997, 94, 13057–13062. [Google Scholar]
- Bertone, P.; Stolc, V.; Royce, T.E.; Rozowsky, J.S.; Urban, A.E.; Zhu, X.; Rinn, J.L.; Tongprasit, W.; Samanta, M.; Weissman, S.; Gerstein, M.; Snyder, M. Global identification of human transcribed sequences with genome tiling arrays. Science 2004, 306, 2242–2246. [Google Scholar]
- Cheng, J.; Kapranov, P.; Drenkow, J.; Dike, S.; Brubaker, S.; Patel, S.; Long, J.; Stern, D.; Tammana, H.; Helt, G.; et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 2005, 308, 1149–1154. [Google Scholar]
- Castle, J.C.; Zhang, C.; Shah, J.K.; Kulkarni, A.V.; Kalsotra, A.; Cooper, T.A.; Johnson, J.M. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008, 40, 1416–1425. [Google Scholar]
- Okoniewski, M.J.; Miller, C.J. Hybridization interactions between probesets in short oligo microarrays lead to spurious correlations. BMC Bioinformatics 2006, 7, 276. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Misquitta, C.; Zhang, W.; Saltzman, A.L.; Mohammad, N.; Babak, T.; Siu, H.; Hughes, T.R.; Morris, Q.D.; et al. Revealing global regulatory features of mammalian alternative splicing using a quantitative microarray platform. Mol. Cell 2004, 16, 929–941. [Google Scholar] [CrossRef]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar]
- Salzberg, S.L. Recent advances in RNA sequence analysis. F1000 Biol. Rep. 2010, 2, 64. [Google Scholar]
- Cloonan, N.; Forrest, A.R.; Kolle, G.; Gardiner, B.B.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar]
- Dinger, M.E. lncRNAs: Finding the forest among the trees? Mol. Ther. 2011, 19, 2109–2111. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Jacquier, A. The complex eukaryotic transcriptome: Unexpected pervasive transcription and novel small RNAs. Nat. Rev. Genet. 2009, 10, 833–844. [Google Scholar] [CrossRef]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [CrossRef]
- Derrien, T.; Guigo, R.; Johnson, R. The long non-coding RNAs: A New (P)layer in the “Dark Matter”. Front Genet. 2011, 2, 107. [Google Scholar]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar]
- Iafrate, A.; Feuk, L.; Rivera, M.; Listewnik, M.; Donahoe, P.; Qi, Y.; Scherer, S.; Lee, C. Detection of large-scale variation in the human genome. Nat Genet. 2004, 36, 949–951. [Google Scholar]
- Sebat, J.; Lakshmi, B.; Troge, J.; Alexander, J.; Young, J.; Lundin, P.; Maner, S.; Massa, H.; Walker, M.; Chi, M.; Navin, N.; Lucito, R.; Healy, J.; Hicks, J.; Ye, K.; Reiner, A.; Gilliam, T.C.; Trask, B.; Patterson, N.; Zetterberg, A.; Wigler, M. Large-scale copy number polymorphism in the human genome. Science 2004, 305, 525–528. [Google Scholar]
- Li, R.; Li, Y.; Zheng, H.; Luo, R.; Zhu, H.; Li, Q.; Qian, W.; Ren, Y.; Tian, G.; Li, J.; et al. Building the sequence map of the human pan-genome. Nat. Biotechnol. 2009, 28, 57–63. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar]
- Kampa, D.; Cheng, J.; Kapranov, P.; Yamanaka, M.; Brubaker, S.; Cawley, S.; Drenkow, J.; Piccolboni, A.; Bekiranov, S.; Helt, G.; et al. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 2004, 14, 331–342. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar]
- Blencowe, B.J. Alternative splicing: New insights from global analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef]
- Mudge, J.M.; Frankish, A.; Fernandez-Banet, J.; Alioto, T.; Derrien, T.; Howald, C.; Reymond, A.; Guigo, R.; Hubbard, T.; Harrow, J. The origins, evolution, and functional potential of alternative splicing in vertebrates. Mol. Biol. Evol. 2011, 28, 2949–2959. [Google Scholar] [CrossRef]
- Ravasi, T.; Suzuki, H.; Pang, K.C.; Katayama, S.; Furuno, M.; Okunishi, R.; Fukuda, S.; Ru, K.; Frith, M.C.; Gongora, M.M.; et al. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res. 2006, 16, 11–19. [Google Scholar]
- Seok, J.; Xu, W.; Jiang, H.; Davis, R.W.; Xiao, W. Knowledge-based reconstruction of mRNA transcripts with short sequencing reads for transcriptome research. PLoS One 2012, 7, e31440. [Google Scholar]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; Kodzius, R.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar]
- Ensembl Genome Browser. Available online: http://useast.ensembl.org/Homo_sapiens/Info/Index (accessed on 5 September 2011).
- NCBI’s RefSeq Database. Available online: http://www.ncbi.nlm.nih.gov/RefSeq/ (accessed on 5 September 2011).
- UCSC Genome Table Browser. Available online: http://genome.ucsc.edu/cgi-bin/hgTables (accessed on 5 September 2011).
- Kapranov, P.; Drenkow, J.; Cheng, J.; Long, J.; Helt, G.; Dike, S.; Gingeras, T.R. Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res. 2005, 15, 987–997. [Google Scholar] [CrossRef]
- Zheng, D.; Frankish, A.; Baertsch, R.; Kapranov, P.; Reymond, A.; Choo, S.W.; Lu, Y.; Denoeud, F.; Antonarakis, S.E.; Snyder, M.; et al. Pseudogenes in the ENCODE regions: Consensus annotation, analysis of transcription, and evolution. Genome Res. 2007, 17, 839–851. [Google Scholar] [CrossRef]
- Sasidharan, R.; Gerstein, M. Genomics: Protein fossils live on as RNA. Nature 2008, 453, 729–731. [Google Scholar] [CrossRef]
- Sie, C.P.; Kuchka, M. RNA editing adds flavor to complexity. Biochemistry (Mosc) 2011, 76, 869–881. [Google Scholar] [CrossRef]
- Bass, B.L.; Weintraub, H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 1988, 55, 1089–1098. [Google Scholar] [CrossRef]
- Wagner, R.W.; Smith, J.E.; Cooperman, B.S.; Nishikura, K. A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc. Natl. Acad. Sci. USA 1989, 86, 2647–2651. [Google Scholar]
- Powell, L.M.; Wallis, S.C.; Pease, R.J.; Edwards, Y.H.; Knott, T.J.; Scott, J. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell 1987, 50, 831–840. [Google Scholar] [CrossRef]
- Chen, S.H.; Habib, G.; Yang, C.Y.; Gu, Z.W.; Lee, B.R.; Weng, S.A.; Silberman, S.R.; Cai, S.J.; Deslypere, J.P.; Rosseneu, M.; et al. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science 1987, 238, 363–366. [Google Scholar]
- Teng, B.; Burant, C.F.; Davidson, N.O. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science 1993, 260, 1816–1819. [Google Scholar]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D.; et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar]
- Li, M.; Wang, I.X.; Li, Y.; Bruzel, A.; Richards, A.L.; Toung, J.M.; Cheung, V.G. Widespread RNA and DNA sequence differences in the human transcriptome. Science 2011, 333, 53–58. [Google Scholar]
- Kleinman, C.L.; Majewski, J. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 2012, 335, 1302. [Google Scholar] [CrossRef]
- Lin, W.; Piskol, R.; Tan, M.H.; Li, J.B. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 2012, 335, 1302-e. [Google Scholar]
- Pickrell, J.K.; Gilad, Y.; Pritchard, J.K. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 2012, 335. [Google Scholar] [CrossRef]
- Schrider, D.R.; Gout, J.F.; Hahn, M.W. Very few RNA and DNA sequence differences in the human transcriptome. PLoS One 2011, 6, e25842. [Google Scholar]
- Barak, M.; Levanon, E.Y.; Eisenberg, E.; Paz, N.; Rechavi, G.; Church, G.M.; Mehr, R. Evidence for large diversity in the human transcriptome created by Alu RNA editing. Nucleic Acids Res. 2009, 37, 6905–6915. [Google Scholar] [CrossRef]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef]
- Costa, V.; Angelini, C.; de Feis, I.; Ciccodicola, A. Uncovering the complexity of transcriptomes with RNA-Seq. J. Biomed. Biotechnol. 2010, 853916. [Google Scholar]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469–477. [Google Scholar]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef]
- Butler, J.; MacCallum, I.; Kleber, M.; Shlyakhter, I.A.; Belmonte, M.K.; Lander, E.S.; Nusbaum, C.; Jaffe, D.B. ALLPATHS: De novo assembly of whole-genome shotgun microreads. Genome Res. 2008, 18, 810–820. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Au, K.F.; Jiang, H.; Lin, L.; Xing, Y.; Wong, W.H. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010, 38, 4570–4578. [Google Scholar] [CrossRef]
- Wang, K.; Singh, D.; Zeng, Z.; Coleman, S.J.; Huang, Y.; Savich, G.L.; He, X.; Mieczkowski, P.; Grimm, S.A.; Perou, C.M.; et al. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010, 38, e178. [Google Scholar]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT--the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; Rinn, J.L.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar]
- Feng, J.; Li, W.; Jiang, T. Inference of isoforms from short sequence reads. J. Comput. Biol. 2011, 18, 305–321. [Google Scholar] [CrossRef]
- Li, W.; Feng, J.; Jiang, T. IsoLasso: A LASSO regression approach to RNA-Seq based transcriptome assembly. J. Comput. Biol. 2011, 18, 1693–1707. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar]
- Oases: De novo transcriptome assembler for very short reads. Available online: http://www.ebi.ac.uk/~zerbino/oases/ (accessed on 12 April 2012).
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar]
- Birol, I.; Jackman, S.D.; Nielsen, C.B.; Qian, J.Q.; Varhol, R.; Stazyk, G.; Morin, R.D.; Zhao, Y.; Hirst, M.; Schein, J.E.; et al. De novo transcriptome assembly with ABySS. Bioinformatics 2009, 25, 2872–2877. [Google Scholar]
- Zhao, Q.Y.; Wang, Y.; Kong, Y.M.; Luo, D.; Li, X.; Hao, P. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: A comparative study. BMC Bioinformatics 2011, 12, S2. [Google Scholar]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [CrossRef]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar]
- Okazaki, Y.; Furuno, M.; Kasukawa, T.; Adachi, J.; Bono, H.; Kondo, S.; Nikaido, I.; Osato, N.; Saito, R.; Suzuki, H.; et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 2002, 420, 563–573. [Google Scholar] [CrossRef]
- Katayama, S.; Tomaru, Y.; Kasukawa, T.; Waki, K.; Nakanishi, M.; Nakamura, M.; Nishida, H.; Yap, C.C.; Suzuki, M.; Kawai, J.; et al. Antisense transcription in the mammalian transcriptome. Science 2005, 309, 1564–1566. [Google Scholar]
- Rinn, J.L.; Euskirchen, G.; Bertone, P.; Martone, R.; Luscombe, N.M.; Hartman, S.; Harrison, P.M.; Nelson, F.K.; Miller, P.; Gerstein, M.; et al. The transcriptional activity of human Chromosome 22. Genes Dev. 2003, 17, 529–540. [Google Scholar]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar]
- Van Bakel, H.; Nislow, C.; Blencowe, B.J.; Hughes, T.R. Most “dark matter” transcripts are associated with known genes. PLoS Biol. 2010, 8, e1000371. [Google Scholar] [CrossRef]
- Asmann, Y.W.; Necela, B.M.; Kalari, K.R.; Hossain, A.; Baker, T.R.; Carr, J.M.; Davis, C.; Getz, J.E.; Hostetter, G.; Li, X.; et al. Detection of redundant fusion transcripts as biomarkers or disease-specific therapeutic targets in breast cancer. Cancer Res. 2012, 72, 1921–1928. [Google Scholar] [CrossRef]
- Clark, M.B.; Amaral, P.P.; Schlesinger, F.J.; Dinger, M.E.; Taft, R.J.; Rinn, J.L.; Ponting, C.P.; Stadler, P.F.; Morris, K.V.; Morillon, A.; et al. The reality of pervasive transcription. PLoS Biol. 2011, 9, e1000625. [Google Scholar] [CrossRef]
- Amaral, P.P.; Mattick, J.S. Noncoding RNA in development. Mamm. Genome 2008, 19, 454–492. [Google Scholar] [CrossRef]
- Berretta, J.; Morillon, A. Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep. 2009, 10, 973–982. [Google Scholar] [CrossRef]
- Kapranov, P.; St Laurent, G.; Raz, T.; Ozsolak, F.; Reynolds, C.P.; Sorensen, P.H.; Reaman, G.; Milos, P.; Arceci, R.J.; Thompson, J.F.; et al. The majority of total nuclear-encoded non-ribosomal RNA in a human cell is ‘dark matter’ un-annotated RNA. BMC Biol. 2010, 8, 149. [Google Scholar] [CrossRef]
- Agarwal, A.; Koppstein, D.; Rozowsky, J.; Sboner, A.; Habegger, L.; Hillier, L.W.; Sasidharan, R.; Reinke, V.; Waterston, R.H.; Gerstein, M. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics 2010, 11, 383. [Google Scholar]
- Malone, J.H.; Oliver, B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011, 9, 34. [Google Scholar] [CrossRef]
- Van Bakel, H.; Nislow, C.; Blencowe, B.J.; Hughes, T.R. Response to “The reality of pervasive transcription”. PLoS Biol. 2011, 9, e1001102. [Google Scholar] [CrossRef]
- Ameur, A.; Zaghlool, A.; Halvardson, J.; Wetterbom, A.; Gyllensten, U.; Cavelier, L.; Feuk, L. Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nat. Struct. Mol. Biol. 2011, 18, 1435–1440. [Google Scholar] [CrossRef]
- Mercer, T.R.; Gerhardt, D.J.; Dinger, M.E.; Crawford, J.; Trapnell, C.; Jeddeloh, J.A.; Mattick, J.S.; Rinn, J.L. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol. 2011, 30, 99–104. [Google Scholar] [CrossRef]
- Jarvis, K.; Robertson, M. The noncoding universe. BMC Biol. 2011, 9, 52. [Google Scholar] [CrossRef]
- Louro, R.; Smirnova, A.S.; Verjovski-Almeida, S. Long intronic noncoding RNA transcription: Expression noise or expression choice? Genomics 2009, 93, 291–298. [Google Scholar]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Dinger, M.E.; Amaral, P.P.; Mercer, T.R.; Pang, K.C.; Bruce, S.J.; Gardiner, B.B.; Askarian-Amiri, M.E.; Ru, K.; Solda, G.; Simons, C.; et al. S. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 2008, 18, 1433–1445. [Google Scholar] [CrossRef]
- Ahituv, N.; Zhu, Y.; Visel, A.; Holt, A.; Afzal, V.; Pennacchio, L.A.; Rubin, E.M. Deletion of ultraconserved elements yields viable mice. PLoS Biol. 2007, 5, e234. [Google Scholar] [CrossRef]
- Monroe, D. Genetics. Genomic clues to DNA treasure sometimes lead nowhere. Science 2009, 325, 142–143. [Google Scholar] [CrossRef]
- Knowles, D.G.; McLysaght, A. Recent de novo origin of human protein-coding genes. Genome Res. 2009, 19, 1752–1759. [Google Scholar] [CrossRef]
- Kaplan, C.D. The architecture of RNA polymerase fidelity. BMC Biol. 2010, 8, 85. [Google Scholar] [CrossRef]
- Ponting, C.P.; Hardison, R. What fraction of the human genome is functional? Genome Res. 2011, 21, 1769–1776. [Google Scholar] [CrossRef]
- Cawley, S.; Bekiranov, S.; Ng, H.H.; Kapranov, P.; Sekinger, E.A.; Kampa, D.; Piccolboni, A.; Sementchenko, V.; Cheng, J.; Williams, A.J.; et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 2004, 116, 499–509. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, J.; Zheng, H.; Li, J.; Liu, D.; Li, H.; Samudrala, R.; Yu, J.; Wong, G.K. Mouse transcriptome: Neutral evolution of ‘non-coding’ complementary DNAs. Nature 2004, 431. [Google Scholar] [CrossRef]
- Pang, K.C.; Frith, M.C.; Mattick, J.S. Rapid evolution of noncoding RNAs: Lack of conservation does not mean lack of function. Trends Genet. 2006, 22, 1–5. [Google Scholar] [CrossRef]
- Ebisuya, M.; Yamamoto, T.; Nakajima, M.; Nishida, E. Ripples from neighbouring transcription. Nat. Cell Biol. 2008, 10, 1106–1113. [Google Scholar] [CrossRef]
- Johnson, J.M.; Edwards, S.; Shoemaker, D.; Schadt, E.E. Dark matter in the genome: Evidence of widespread transcription detected by microarray tiling experiments. Trends Genet. 2005, 21, 93–102. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pertea, M. The Human Transcriptome: An Unfinished Story. Genes 2012, 3, 344-360. https://doi.org/10.3390/genes3030344
Pertea M. The Human Transcriptome: An Unfinished Story. Genes. 2012; 3(3):344-360. https://doi.org/10.3390/genes3030344
Chicago/Turabian StylePertea, Mihaela. 2012. "The Human Transcriptome: An Unfinished Story" Genes 3, no. 3: 344-360. https://doi.org/10.3390/genes3030344
APA StylePertea, M. (2012). The Human Transcriptome: An Unfinished Story. Genes, 3(3), 344-360. https://doi.org/10.3390/genes3030344