Abstract
The entire publicly available set of 37 genome sequences from the bacterial order Chlamydiales has been subjected to comparative analysis in order to reveal the salient features of this pangenome and its evolutionary history. Over 2,000 protein families are detected across multiple species, with a distribution consistent to other studied pangenomes. Of these, there are 180 protein families with multiple members, 312 families with exactly 37 members corresponding to core genes, 428 families with peripheral genes with varying taxonomic distribution and finally 1,125 smaller families. The fact that, even for smaller genomes of Chlamydiales, core genes represent over a quarter of the average protein complement, signifies a certain degree of structural stability, given the wide range of phylogenetic relationships within the group. In addition, the propagation of a corpus of manually curated annotations within the discovered core families reveals key functional properties, reflecting a coherent repertoire of cellular capabilities for Chlamydiales. We further investigate over 2,000 genes without homologs in the pangenome and discover two new protein sequence domains. Our results, supported by the genome-based phylogeny for this group, are fully consistent with previous analyses and current knowledge, and point to future research directions towards a better understanding of the structural and functional properties of Chlamydiales.
1. Introduction
Members of the order Chlamydiales are obligate intracellular bacteria, characterized by a unique developmental cycle and are important pathogens of humans and animals resulting in a wide range of diseases, including several zoonoses [1,2,3]. The order Chlamydiales, separated from other eubacteria by forming a deep branch in ribosomal RNA-based phylogenetic trees, has been enriched by new lineages. Beside the family Chlamydiaceae, in which important chlamydial pathogens are grouped, new families, such as Parachlamydiaceae, Simkaniaceae and Waddliaceae, have been recognized to accommodate newly discovered pathogenic and non-pathogenic chlamydial organisms [4,5,6].
Since the release of the first chlamydial genome sequence from Chlamydia trachomatis (serovar D) [7], new genomes are being sequenced, thus offering insights into the genome organization and functional capacity of the corresponding species [8]. Besides its crucial importance for applied research in medical and veterinary microbiology [9], this corpus of genomic information is also key to understanding the evolutionary position of various chlamydial species (or strains) and the inference of the internal phylogeny of this distinct taxon [8,10,11,12].
As the intracellular lifestyle imposes constraints on gene content and metabolic capabilities, the Chlamydiales might represent one of the best datasets for the development of pangenome analysis methods [13]. Additional challenges are the wide variety of chlamydial genome sizes with unequal rates of reduction, and a repertoire of less characterized proteins than other bacterial groups whose pangenomes have been analyzed, e.g., Streptococcus or Salmonella [14,15].
Previously, we have used the genome of Chlamydia trachomatis [7] as a case study for annotation transfer quality [16]. Using a novel encoding scheme and a scoring function called TABS for transitive annotation-based scale [16], our main finding regarding annotation was that, despite a number of inconsistencies, automated annotation pipelines performed remarkably well when benchmarked against a manually curated annotation corpus [16]. These results are important for the quantification of reproducibility and consistency in genome-wide annotation [17].
In this work, we explore the entire set of the Chlamydiales pangenome with a broad collection of genome sequences publicly available to date (31 Chlamydiaceae and six other Chlamydiales genomes), twice as many as in a similar recent analysis [18]. Importantly, our pangenome analysis pipeline incorporates recently sequenced genomes of key Chlamydiaceae species not previously reported, thus augmenting our understanding from previous findings [18,19].
We focus on key aspects of pangenome analysis and explore multiple facets of the Chlamydiales gene content in terms of protein-coding genes and families. We also provide certain key findings that might illuminate the evolutionary history of this group as well as interesting sequence motifs not widely shared within this order. Beyond the confirmation of the recent analysis of the Chlamydiales as mentioned above [18], we also use this group to expand on methods for pangenome analysis [13,20] by proposing a pangenome analysis pipeline. Our results are consistent with wider studies of pangenomes [21] and provide additional knowledge for Chlamydiales. In conclusion, pangenome analysis offers an opportunity for the study of bacterial genome evolution, the development of relevant methods and the understanding of genome structure and proteome function on a large scale.
2. Experimental Section
2.1. Data Collection
All protein sequence data from 37 genomes were compiled into a single data collection (February–July 2011), including the most recent published Chlamydiales genomes. In total, 43,736 protein-coding genes were extracted from public databases corresponding to the entire set of 37 genome sequences from the bacterial order Chlamydiales currently available (Table 1). Sequence data were codified following the style of the COGENT database [22], for easy identification both by programs and human users (Supplement S1). The above notation is followed throughout this work. The COGENT scheme encodes genus and species names into a four-character identifier prefix string, followed by a code for the strain name, its version (in this collection all versions are considered as version 1 and optionally hidden) and finally for proteins the relative order of the sequence within the genome [23] (Table 1). We have also recorded the date of publication for the corresponding genome (or the release date where no publication was available) (Supplement S2).

Table 1.
List of Chlamydiales genome sequences used in this study.
| ## | Species and Strain Name/Codes | Internal Identifier | Protein-Coding Genes |
|---|---|---|---|
| 01 | Candidatus Protochlamydia amoebophila UWE25 | CPRO-UWE-01 | 2,031 |
| 02 | Chlamydia muridarum Nigg | CMUR-NIG-01 | 911 |
| 03 | Chlamydia trachomatis 434/Bu | CTRA-434-01 | 874 |
| 04 | Chlamydia trachomatis A/HAR-13 | CTRA-AHA-01 | 919 |
| 05 | Chlamydia trachomatis B/Jali20/OT | CTRA-BJA-01 | 875 |
| 06 | Chlamydia trachomatis B/TZ1A828/OT | CTRA-BTZ-01 | 880 |
| 07 | Chlamydia trachomatis D/UW-3/CX | CTRA-DUW-01 | 895 |
| 08 | Chlamydia trachomatis L2b/UCH-1/proctitis | CTRA-L2B-01 | 874 |
| 09 | Chlamydophila abortus S26/3 | CABO-S26-01 | 932 |
| 10 | Chlamydophila caviae GPIC | CCAV-GPI-01 | 1,005 |
| 11 | Chlamydophila felis Fe/C-56 | CFEL-FEC-01 | 1,013 |
| 12 | Chlamydophila pneumoniae AR39 | CPNE-AR3-01 | 1,112 |
| 13 | Chlamydophila pneumoniae CWL029 | CPNE-CWL-01 | 1,052 |
| 14 | Chlamydophila pneumoniae J138 | CPNE-J13-01 | 1,069 |
| 15 | Chlamydophila pneumoniae TW-183 | CPNE-TW1-01 | 1,113 |
| 16 | Waddlia chondrophila WSU 86-1044 | WCHO-WSU-01 | 1,956 |
| 17 | Chlamydia trachomatis E/150 | CTRA-E15-01 | 927 |
| 18 | Chlamydophila pecorum E58 | CPEC-E58-01 | 988 |
| 19 | Chlamydophila psittaci 6BC | CPSI-6BC-01 | 975 |
| 20 | Chlamydophila abortus LLG | CABO-LLG-01 | 925 |
| 21 | Chlamydophila pneumoniae LPCoLN | CPNE-LPC-01 | 1,105 |
| 22 | Chlamydophila psittaci Cal10 | CPSI-CAL-01 | 1,005 |
| 23 | Parachlamydia acanthamoebae UV7 | PACA-UV7-01 | 2,788 |
| 24 | Parachlamydia acanthamoebae str. Hall’s coccus | PACA-HAL-01 | 2,809 |
| 25 | Simkania negevensis Z | SNEG-ZXX-01 | 2,518 |
| 26 | Waddlia chondrophila 2032/99 | WCHO-203-01 | 2,015 |
| 27 | Chlamydophila psittaci 01DC11 | CPSI-01D-01 | 975 |
| 28 | Chlamydophila psittaci 02DC15 | CPSI-02D-01 | 978 |
| 29 | Chlamydophila psittaci 08DC60 | CPSI-08D-01 | 973 |
| 30 | Chlamydia trachomatis D-EC | CTRA-DEC-01 | 878 |
| 31 | Chlamydia trachomatis D-LC | CTRA-DLC-01 | 878 |
| 32 | Chlamydia trachomatis E/11023 | CTRA-E11-01 | 926 |
| 33 | Chlamydia trachomatis G/11074 | CTRA-G74-01 | 919 |
| 34 | Chlamydia trachomatis G/11222 | CTRA-G22-01 | 927 |
| 35 | Chlamydia trachomatis G/9301 | CTRA-G93-01 | 921 |
| 36 | Chlamydia trachomatis G/9768 | CTRA-G97-01 | 920 |
| 37 | Chlamydia trachomatis Sweden2 | CTRA-SWE-01 | 875 |
| Total | 43,736 |
The first column signifies the inclusion order into the genome collection and does not reflect any other relationship. The second column lists the species and strain name, the third column the COGENT-style identifier and the last column the number of protein-coding genes.
2.2. Sequence Comparison
All protein sequence data were masked using CAST with default parameters (threshold = 40), to exclude compositionally biased regions [24]. In total, 6,906 such regions were filtered out, provided for further study (Supplement S3).
The masked sequences were used as queries against the genome corpus, in an all-against-all mode with BLAST (blastall, e-value threshold 10−6) [25,26]; in total, more than 40,000 BLAST searches were performed and 1,709,325 significant similarities below threshold were obtained (Supplement S4).
2.3. Clustering and Annotation
The similarity pairwise list (from Supplement S4) was submitted to MCL sequence clustering [27], with default parameters (e.g., inflation value 2.0); clusters were incrementally assigned to an integer identifier. Clusters are sorted by their size (number of members in a cluster, Supplement S5); thus, the largest clusters have smallest-integer identifiers (see Results and Appendix-Table 1). This approach has also been used successfully elsewhere [28] as a method of choice.
Annotation transfer based on the first chlamydial genome ever sequenced was implemented through the direct matching of the lead sequences to a previously highly curated dataset for Chlamydia trachomatis D/UW-3/CX [7].
The annotation qualifiers used in the manually curated corpus [16] are: ENZYME (for enzymes with EC number assignments), FUNCTION (for other protein functions), SIMILAR-TO (for those sequences with a similarity to a protein of known function but no specific assignment) and DOMAIN (for the existence of a known, named protein sequence domain) [16] (Appendix-Table 1).
Sequence matching of the original dataset to the data collection presented here was performed by MagicMatch [29], which was the first scheme to implement the MD5 checksum for protein sequence identification, an approach later propagated in all major database resources.
2.4. Analysis of Unique Genes
All unique genes, i.e., more than 2,000 genes with no similarity within the pangenome, were searched against the non-redundant protein sequence database (nrdb: 15,052,178 entries) [30]. Results from this search were evaluated manually and key similarities were extracted for further investigation (Supplement S6).
2.5. Genome Trees
Genome-based trees were calculated using phylogenetic profile distance [31,32]. Similarity values were measured by the shared number of genes represented by phylogenetic profiles, symmetrified by the minimum shared value, normalized by minimum self-similarity and turned into distance values as previously described [32,33] (Supplement S7).
2.6. Sequence Alignments
Multiple sequence alignments were performed and visualized by JalView [34]. Novel motifs reported in this work are provided below and in FASTA format (Supplements S8,9).
2.7. Data Availability
Per genome contributions to the pangenome are also provided (Supplement S10). All sequence data and results (in 10 Supplements) have been made available at datadryad.org, under the identifier [35].
3. Results
3.1. General Characteristics of the Chlamydiales Pangenome
The Chlamydiales collection herein contains over 40,000 protein-coding genes in total, with ~1,200 genes/genome on average, with significant deviations (Table 1). We take the view to present the two extreme tails of this data collection in detail following the clustering step for the identification of protein families within the pangenome and comment on the intermediate cases. In other words, we primarily focus on the two classes of the most interesting clusters, (i) those containing the core genes and (ii) those corresponding to “unique” genes, without significant similarities within the pangenome, thus singleton clusters. The functional characterization of the entire complement as well as further issues listed in the discussion for future research are clearly beyond the scope of this critical review.
3.2. Protein Families
In total, the clustering has yielded 5,554 clusters corresponding to protein families. For practical purposes, we define a protein family as one that contains at least three genes: in that sense, there are 294 cases, which do not detect themselves in this comparison (typically because of either short length, abnormal composition, or both), 2,038 unique genes (singletons) and 1,177 doublets. The remaining 2,045 clusters represent protein families with three or more members, distributed across 37 genomes (Figure 1).
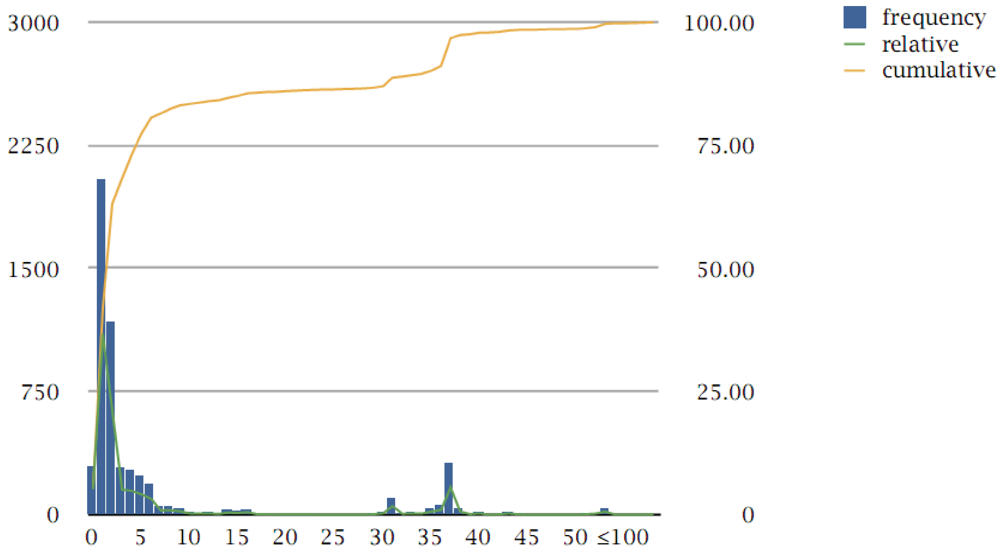
Figure 1.
Pangenome protein family size distribution. Cluster size is displayed on the x-axis (bins until 50 are all shown; above 50, bins are shown for each ten counts, labels for every five bin sizes); absolute frequency of clusters is shown on the left y-axis (bars, green curve); cumulative count of clusters is shown on the right y-axis (orange curve). Families are defined as those clusters with at least three members (see text); all cluster frequencies are shown here for completeness. The bimodal nature of the distribution can be seen between the peak at low cluster sizes and 37; above 37 there are multi-member and multi-species protein families (see text).
It is evident that the protein family size distribution follows, as expected, the shape of other pangenome analyses, with a clear bimodal distribution, with one peak at low-count families which has been called the “accessory pool” and another peak at the limit of the genomes under consideration, which has been called the “extended core” [21]. The so-called “character genes” (which we prefer to define as “peripheral”, as opposed to “core” genes) exhibit, by definition, a heterogeneous distribution across genomes (and between peaks) and present an additional challenge for further interpretation (Figure 1).
The peak at exactly 37 with 312 counts, i.e., 312 families with exactly 37 members, corresponds to the number of 37 genomes analyzed across the pangenome. Beyond that peak, there are 180 protein families with more than 37 members (clusters 1–180) (Supplement S5), of which ten contain more than 100 members and are discussed below.
3.3. Multi-Member Families
The four largest families with more than 120 members are represented by the ABC transporter permeases (530 members), the polymorphic outer membrane proteins of Chlamydiaceae [36] (POMPs, 435 members), the flagellum-specific ATP synthases/type III secretion system ATPases, e.g., CT669 [37] (152 members), and a family of unknown function recently characterized as type III secreted effectors [38] (DUF582, 140 members) (Figure 2).
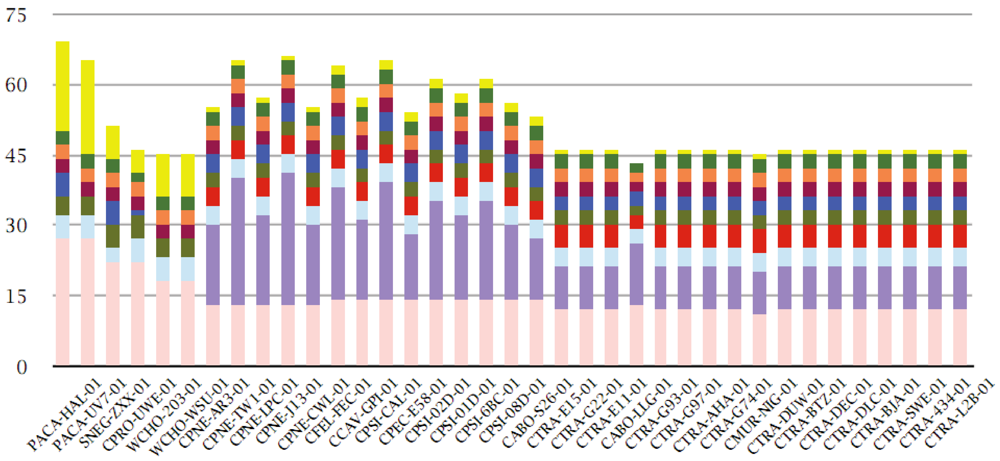
Figure 2.
Top ten multi-member families within the pangenome. Genomes (with full COGENT-like codes) are shown on the x-axis, sorted by total protein-coding gene count (see also Table 1). Absolute cumulative counts of multi-member families are shown on the y-axis (displayed in the figure legend from left to right and then top to bottom, e.g., ABC transporter permeases, POMPs, type III secretion system ATPases, etc. according to size, see text), color coded according to figure legend.
Following those, there are another four families with more than 110 members each: the EF-Tu/EF-G/LepA family (119 members), the oligopeptide binding protein family OppA (114 members), the GroEL family (111 members) and finally the Ile-Leu-Val (ILV)-tRNA synthetases (111 members). These are followed by two families with more than 100 members, namely the Dihydrolipoamide acetyltransferase E2 component/Dihydrolipoamide succinyltransferase (110 members) and the 3-oxoacyl-[acyl-carrier protein] reductase families (109 members) (Figure 2).
A significant number of multi-member families contain proteins of known function (Supplement S5). Interestingly, families containing only homologues from S. negevensis, W. chondrophila, P. acanthamoebae and Protochlamydia amoebophila are 172 in total, remarkably close to the 171 clusters of “orthologous” proteins in this group of species reported recently [18].
3.4. Core Genes
At the other end of the bimodal distribution, there are 312 families with 37 genes each, reflecting the number of genomes analyzed. However, there are eight clusters here with duplicates per genome (clusters 224, 460: S. negevensis; 254, 276, 420: P. acanthamoebae; 255, 272: P. amoebophila; 429: W. chondrophila 203) (two of which of unknown functional roles, Appendix-Table 1). Thus, there are exactly 304 protein families with 37 genes each represented once in each genome, which can be truly called “core” genes, most of which have some source of annotation (Appendix-Table 1). These represent just over a quarter of the average chlamydial genome (304/1182 = 26%).
Annotations transferred from the manually curated seed annotation corpus of C. trachomatis reveal a wide range of functional roles for this core set, as expected (Appendix-Table 1). Indeed, 227 families of the core set can be assigned to a functional role, according to the annotation qualifiers originally used (see Experimental Section). Only an additional 77 cases in this set do not contain any annotation (Appendix-Table 1). It can be argued, therefore, that this level of characterization of 75% (227/304) across 37 genomes signifies a functional coherence that is consistent with our current knowledge of this taxonomic order. This list is provided for further investigation by the community; it is worth pointing out that it encompasses basic cellular roles in genetic information processing (e.g., cluster 184), including transcription (e.g., cluster 187) and translation (e.g., clusters 242–243), metabolic transformations (e.g., cluster 182 or 196), transport systems (e.g., clusters 193–195) and other key processes (e.g., cluster 192). It is interesting to note that apart from complements represented by ribosomal proteins or aminoacyl-tRNA synthetases, other systems are also coherently detected, for example the NifU [39]/NifS [40] genes (clusters 221–222).
3.5. Peripheral Genes
In the midst of the two extremes (viz. peaks) of the bimodal family size distribution, there exists a wide variety of cases with an anomalous and clearly heterogeneous pattern. There are 428 families with more than ten and less than 37 members (not shown, available in Supplement S5). Their hererogeneous composition is reflected by the fact that 217 of the 428 families (just over 50%) do not contain a homolog outside the Chlamydiaceae, i.e., across the larger genomes mentioned above. Within this group, however, there is a significant variation of family phylogenetic distribution (not shown) that needs to be explored in future research.
3.6. Unique Genes
In total, there are 2,038 unique genes represented by singleton clusters, thus not falling into families within the pangenome. The content of genomes with unique genes varies significantly, from 0 to 796 (S. negevensis), with 55 unique genes on average. In percentage points, this varies from obviously 0 to 32% of the genome (S. negevensis), with an average of just over 3% per genome (Figure 3).
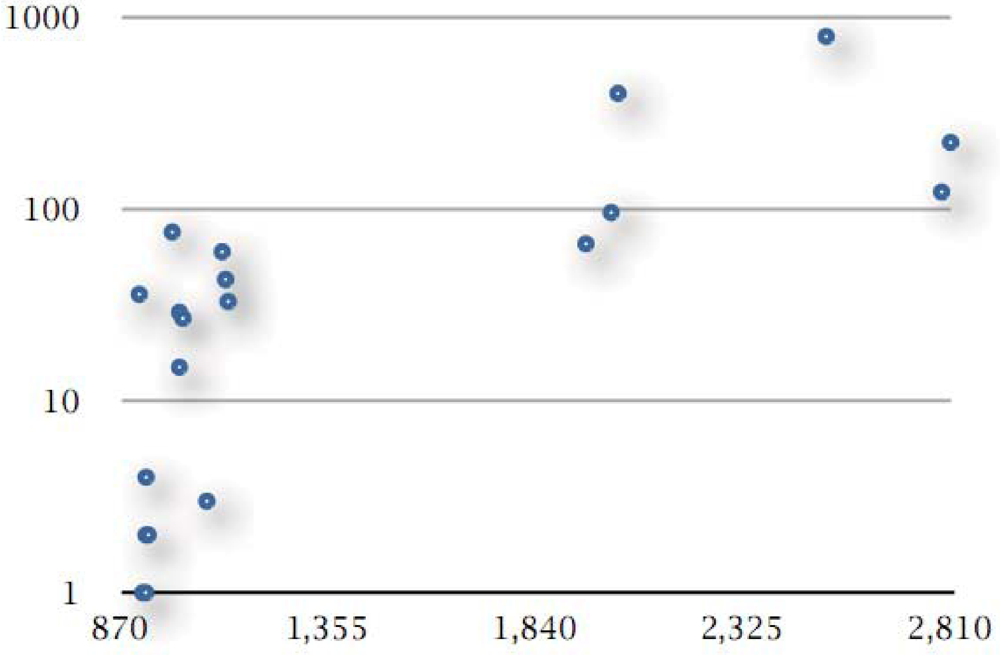
Figure 3.
Correlation between genome size and unique genes. Genome size is given as the number of protein-coding genes (shown on the x-axis) against the count of unique genes (number of unique genes without homologs within the pangenome, shown on the y-axis; y-axis is displayed on logarithmic scale). The six points on the upper right part of the graph are evidently those genomes with largest gene counts, all outside the Chlamydiaceae family (see Table 1 and text). The pattern observed is primarily due to the sampling of taxonomic space of the Chlamydiales and will vary as more genomes from this group become available.
The densest part of the phylogeny exhibits no unique genes—17 genomes, including most of the C. trachomatis and C. psittaci strains, C. pneumoniae CWL029 and C. abortus LLG (Figure 3, missing points corresponding to 17 genomes with zero value on the y-coordinate, available in Supplement S5). Twenty genomes have unique genes, of which six genomes have less than 10 such genes and one with 15 unique genes (Figure 3), all from the above group, or less than 2% of their genome entries. Another five genomes with a handful of unique genes are C. pneumoniae AR39 (33/3%), TW-183 (43/4%) and LPCoLN (60/5%) as well as C. felis (27/3%) and C. caviae (29/3%). The remaining eight genomes contain the majority of unique genes, 1818 in number or 89% of total, ranging from 66 (W. chondrophila WSU, 3% of genome) to 796 genes (S. negevensis, 32% of genome). This is not entirely a biological effect, rather a sampling artifact arising from the deeper sequencing of the C. trachomatis/C. pneumoniae group (see below).
The six outliers which form a different group above (upper right, Figure 3) are all species with large genomes (ca. 2,000 protein-coding genes or more): the two W. chondrophila strains (3–4%), the two P. acanthamoebae strains (4–8%), P. amoebophila (20%), and S. negevensis (32%), listed here according to the absolute number of their unique genes per genome. In relative terms, however, two species namely C. muridarum (36/4%), and C. pecorum (76/8%) contain a significant number of unique genes given their relatively small genome size (both less than 1,000 protein-coding genes).
3.7. Properties of Unique Genes
The genes considered as singletons in this analysis are 2,038 as mentioned above. Of those, a number of short genes might fall into pangenome families (not shown) but do not seriously affect the overall assessment (e.g., case CCAV-GPI-01-000824 in Supplement S6). This is an artifact of sensitivity for the two different searches, first against the 40,000 or so genes of the pangenome and second against the entire nrdb database of more than 15 million sequences. While a full analysis of the unique gene complement of the Chlamydiales is under progress, it is interesting to report on a number of findings pertinent to this work.
A number of genes from the pangenome have identified homologs such as cell-wall associated hydrolases (TC0114 from C. muridarum Nigg), proteins of unknown function (e.g., pc0061, pc0549, pc0850, pc0855), endonucleases (e.g., pc0252), exonucleases (pc0951), transposases (e.g., pc0068), DNA repair proteins (e.g., pc0286), acyltransferases (e.g., pc0180), Mg chelatases (pc0480), oxidoreductases (pc0504), streptomycin 6-kinases (e.g., pc0510), metallophosphoesterases (pc0948) from P. amoebophila and LmbE/ypjG family proteins (e.g., wcw_0275) or transposases (e.g., wcw_0482) from W. chondrophila WSU. Similarly, multiple cases of similarity to families of known or unknown function are discovered for unique genes from the larger genomes (not shown).
One such domain is an enigmatic, short and highly conserved motif containing the triplet Pro-Cys-Tyr (PCY), present in the C. pneumoniae AR39 CP0988 protein. This protein is 52 residues long and does not exhibit significant similarities to any other protein in the Chlamydiales pangenome. However, it does show similarity to a set of short proteins (<100 residues long) from various species, including Acinetobacter, Brucella, Clostridium, Coxiella, Curvibacter, Eubacterium, Parvimonas, Rhizobium, Ruminococcus, Selenomonas, Streptomyces, other longer proteins from Chloroflexi, Heliobacterium, Lactobacillus, the C-terminus of a Propionibacterium protein (HL046PA2) and an uncultured Acidobacteria bacterium HF4000_26D02, and importantly, to a number of longer plant proteins from Nicotiana tabacum, Pinus koraiensis, Solanum demissum (middle of protein) and Vitis vinifera (N-terminus, total length 1,193 residues) (Supplement S8). This conserved region with this peculiar phylogenetic distribution has not been characterized previously to our knowledge, and can be considered a genuine novel domain of unknown function (Figure 4). It remains unclear whether the domain has been universally lost from the Chlamydiales pangenome or acquired from C. pneumoniae through horizontal transfer.
Another interesting example of a unique protein is the P. amoebophila pc0506. This 82-residue-long uncharacterized protein is evidently absent from the core pangenome and yet it exhibits significant similarity to four Verrucomicrobia proteins from Verrucomicrobium spinosum, Chthoniobacter flavus, Pedosphaera parvula and Coraliomargarita akajimensis, in this order of similarity, ranging from 53% down to 44% sequence identity (Figure 5). The above mentioned proteins reportedly belong the leucyl aminopeptidase superfamily (Supplement S9). The functional significance of this biochemical role for P. amoebophila is not yet understood. Yet, the strong mutual similarity of this protein family with Verrucomicrobial and P. amoebophila members (no other member in the entire pangenome) can be placed within the general controversy of the connection of Chlamydiales with the so-called PVC group [41,42] (see below).

Figure 4.
Alignment of the PCY domain. The PCY motif is centered around position 15 of the multiple alignment. The domain was discovered following five iterations with PSI-BLAST with CP0988 as query sequence (GI:16752158), until convergence and an e-value threshold 0.005. In total 70 sequences were recovered; redundancy was removed at 95% with Jalview [34], resulting in 32 sequences shown here. The length of the domain is just 30 residues; boxes signify sequence identity at 50% or above (darker color: more conserved). GI labels are provided, along with sequence coordinates on the left of the alignment (see text for more details and discussion).
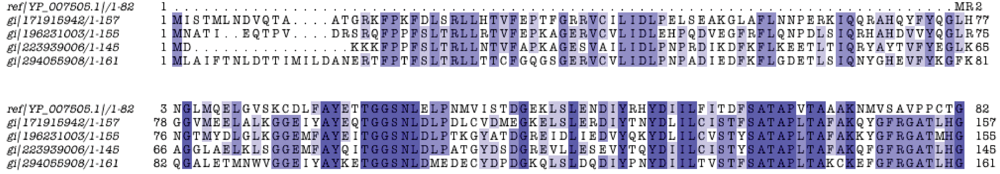
Figure 5.
Alignment of a unique leucyl aminopeptidase family. The domain was discovered following five iterations with PSI-BLAST with pc0506 as query sequence (YP_007505.1). Display conventions as in Figure 4.
In all, it appears that properties encoded from most unique genes, apart from their unusual phylogenetic distribution, represent accessory functional roles that provide additional versatility to the largest genomes in the group, possibly related to their extra functional capabilities. Two exceptions with seemingly central functions are wcw_0805, with similarity to the 50S L34 ribosomal protein family and wcw_861, with similarity to 6-pyruvoyl tetrahydrobiopterin synthases, both from W. chondrophila WSU (not shown).
3.8. Protein Family Contributions from Genome Projects
As mentioned above, we have tracked the original publication (and/or release) data for the genomes under consideration, in terms of novel families detected per genome sequence (Supplement S10). By mapping the protein families which appear first in this ranking order, we can thus estimate the relative “novelty” or contribution of previously unseen protein families within the chlamydial pangenome and the typical “pangenome saturation curve” (Figure 6).
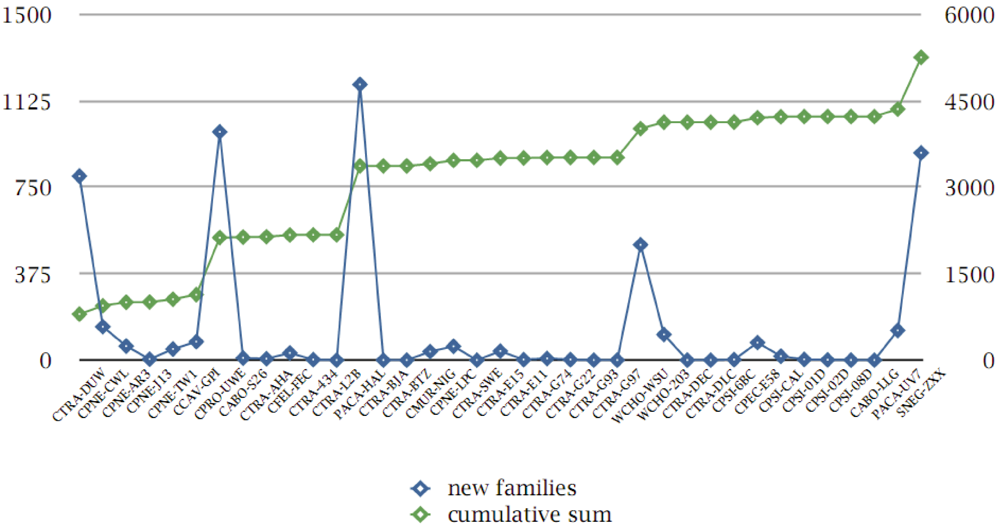
Figure 6.
Protein family contributions from genome projects. Genome codes are sorted according to their original publication date (and/or release date, x-axis); absolute number of “novel” protein families within the pangenome are given (left y-axis, blue curve and square symbols); cumulative sum of protein families (up to 5,260, excluding those without self-hits, see text) is also shown, defined as a “pangenome saturation curve” (right y-axis, green curve and square symbols).
As expected, and discussed above (Figure 3), for the densest part of the group, little or no contributions have been provided. Apart from the larger genomes, which have added hundreds of new gene types [19], the more distant members of the group with small genomes, for instance C. caviae or C. pecorum, have also contributed a significant number (80 and 76, respectively—Supplement S10).
3.9. Genome Phylogeny
Finally, we have reconstructed the genome phylogeny of the pangenome based on the sharing of phylogenetic profile patterns based on the above analysis (see Experimental Section). Evidently, the pangenome is stratified according to the known, established phylogeny patterns [10] (Figure 7). The genome tree is another concise way to visualize the “novelty” components of the various species and strains that have been sequenced, exemplified above in various contexts, e.g., number of unique genes (Figure 3) or the tracking of the relative contributions of novel protein families (Figure 6). A future aspect of this work will be to infer the history of the pangenome using methods of ancestral state reconstruction [43]. The evolutionary history of the Chlamydiales as reflected by the genome tree might also shed light on the ongoing controversy about their status within the tree of life [41].
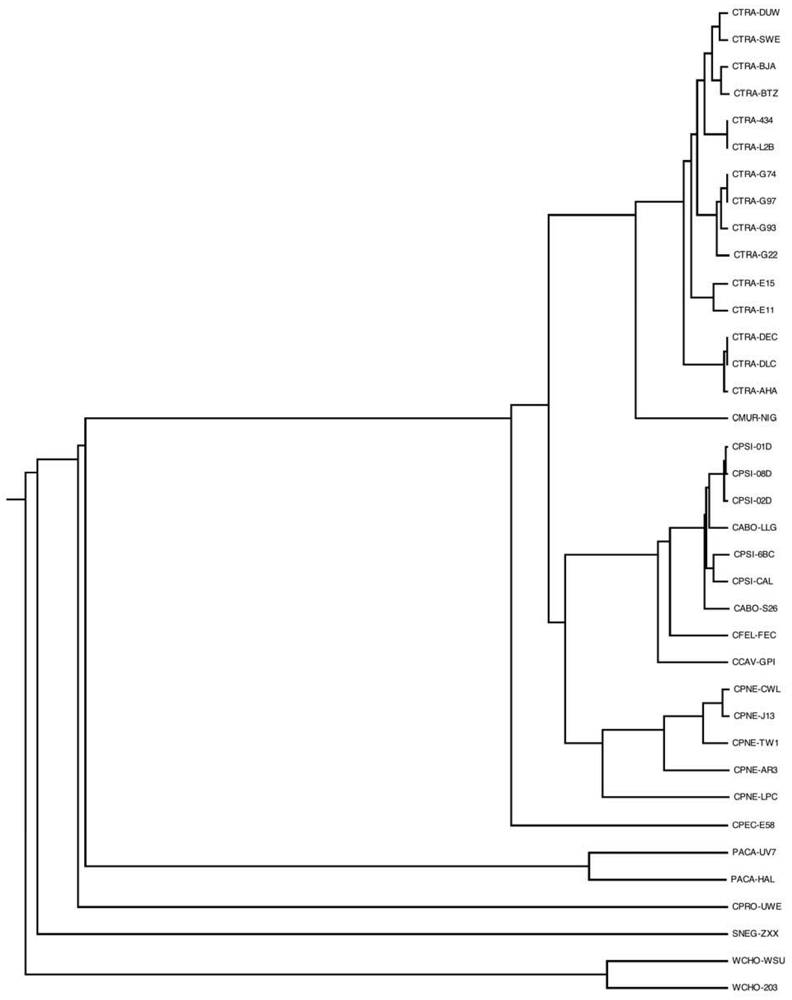
Figure 7.
Genome tree of the Chlamydiales. Dendrogram representing phylogenetic relationships of the 37 Chlamydiales genomes analyzed, based on sharing of phylogenetic profiles (see Experimental Section for details). Genome codes are given as labels.
The genome tree accurately reflects the current taxonomy of Chlamydiales [4,44], with a couple of notable exceptions namely the clustering of C. abortus with C. psittaci, the closer relationship of C. felis with the former two species against C. caviae—in agreement with previous findings [6,44] but not with other proposals [8]—as well as the distinct relationship of C. pecorum at the root of Chlamydiacae and not as a sister group of C. pneumoniae [4,44]. The resulting phylogenetic tree using genome-wide phylogenetic profile sharing patterns can also act as an internal control of the pangenome analysis, since all the closely related strains sequenced are grouped together with very high accuracy (Figure 7).
4. Discussion
Our results suggest that the Chlamydiales pangenome reflects a certain degree of structural stability, as core genes represent over a quarter of an average genome, as well as functional coherence, in the sense that most functional properties of these genes are consistent with current knowledge. Unlike various claims in the recent literature, it turns out that, at least in the case of a highly constrained pangenome of intracellular pathogens, there is an unexpected degree of stability, given the wide range of phylogenetic relationships within this particular taxon.
It is thus shown that for the smallest of genomes (<900 protein-coding genes), over a third of their gene content is shared with larger genomes (>2,000 genes), decorated by a broader element of so-called “character”, or peripheral, genes. This distribution, which in turn is influenced by the sampling of phylogeny and other factors, requires further investigation, being beyond the scope of this work.
It should also be pointed out that the Chlamydiales pangenome exhibits general characteristics of distribution not dissimilar to other recent pangenome analyses, including those of the Salmonella pangenome with 45 strains [15], the Streptococcus pneumoniae pangenome with 44 strains [14] and the Campylobacter pangenome with 96 strains [28], suggesting the conservation of a core pangenome within and across bacterial taxa that have been sampled adequately. In the case of Salmonella, tracking the contributions of new strains to the entire core set and the pangenome suggests a slight expansion with more sampling and a stable core, reminiscent of the Chlamydiales, with one third of the pangenome represented in the core set [15]. A slightly less stable pattern is detected in the Streptococcus pneumoniae group [14], possibly due to a wider diversity in that sample, yet with a similar pattern of core set saturation. Interestingly, an attempt for ancestral reconstruction in the S. pneumoniae/S. mitis complex suggests that there is a dual process of genome expansion and reduction in the different paths leading to the genomes of contemporary strains [14]. A more comprehensive analysis of the Campylobacter pangenome with 96 strains [28], using a combination of experimental and theoretical work, also points to the same direction: Within the two species groups examined, the core gene set overlap reaches 80%, supporting earlier findings for the related Helicobacter pylori strains [45].
5. Conclusions
We have thus examined the salient features of the Chlamydiales pangenome, introducing a pangenome analysis pipeline and certain definitions that facilitate the discovery of core and peripheral genes, the identification of unique genes with various origins as well as the detection of novel protein sequence domains. We expect that analogous efforts will lead to rigorous standards for pangenome analysis in the future. Future research opportunities abound, for example: ancestral reconstruction [43], syntenic patterns of genome structure (e.g., [28,45]), the (presently limited) enrichment with expression data, the evolutionary histories of ‘peripheral’ genes (as discussed above), the connection of Chlamydiales with plants [46,47,48,49,50], the position of the Chlamydiales in the tree of life, and the connection with the PVC superphylum [41,42,50]. Wider challenges that go beyond the above pangenome-specific issues might include a more detailed annotation of the entire dynamic range of family distribution [21], the characterization of protein function in a wider context including comparative metabolic reconstructions [19], the evolution of mobile elements [51], the deeper understanding of the physiological and pathological properties [52,53] of the strains that have been sequenced and the connection with other pangenomes [28].
Acknowledgements
Parts of this work have been supported by the FP6 Network of Excellence ENFIN (contract # LSHG-CT-2005-518254) and the FP7 Collaborative Project MICROME (grant agreement # 222886-2), both funded by the European Commission. C.A.O. thanks the Department of Biological Sciences at the University of Cyprus for their kind hospitality during the spring semester of 2012.
References
- Wyrick, P.B. Intracellular survival by Chlamydia. Cell. Microbiol. 2000, 2, 275–282. [Google Scholar]
- Corsaro, D.; Venditti, D. Emerging chlamydial infections. Crit. Rev. Microbiol. 2004, 30, 75–106. [Google Scholar]
- Horn, M. Chlamydiae as symbionts in eukaryotes. Annu. Rev. Microbiol. 2008, 62, 113–131. [Google Scholar]
- Everett, K.D.; Bush, R.M.; Andersen, A.A. Emended description of the order Chlamydiales, proposal of Parachlamydiaceae fam. nov. and Simkaniaceae fam. nov., each containing one monotypic genus, revised taxonomy of the family Chlamydiaceae, including a new genus and five new species, and standards for the identification of organisms. Int. J. Syst. Bacteriol. 1999, 49, 415–440. [Google Scholar]
- Ludwig, W.; Euzéby, J.; Whitman, W.B. Road map of the phyla Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes. In Bergey’s Manual of Systematic Bacteriology, 2nd; Krieg, N.R., Staley, J.T., Brown, D.R., Hedlund, B.P., Paster, B.J., Ward, N.L., Ludwig, W., Whitman, W.B., Eds.; Springer-Verlag: New York, NY, USA, 2010; Volume 4, pp. 1–19. [Google Scholar]
- Corsaro, D.; Valassina, M.; Venditti, D. Increasing diversity within Chlamydiae. Crit. Rev. Microbiol. 2003, 29, 37–78. [Google Scholar]
- Stephens, R.S.; Kalman, S.; Lammel, C.; Fan, J.; Marathe, R.; Aravind, L.; Mitchell, W.; Olinger, L.; Tatusov, R.L.; Zhao, Q.; et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 1998, 282, 754–759. [Google Scholar]
- Stephens, R.S.; Myers, G.; Eppinger, M.; Bavoil, P.M. Divergence without difference: Phylogenetics and taxonomy of Chlamydia resolved. FEMS Immunol. Med. Microbiol. 2009, 55, 115–119. [Google Scholar]
- Wang, Y.; Kahane, S.; Cutcliffe, L.T.; Skilton, R.J.; Lambden, P.R.; Clarke, I.N. Development of a transformation system for Chlamydia trachomatis: Restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog. 2011, 7, e1002258. [Google Scholar]
- Horn, M.; Collingro, A.; Schmitz-Esser, S.; Beier, C.L.; Purkhold, U.; Fartmann, B.; Brandt, P.; Nyakatura, G.J.; Droege, M.; Frishman, D.; et al. Illuminating the evolutionary history of chlamydiae. Science 2004, 304, 728–730. [Google Scholar]
- Subtil, A.; Dautry-Varsat, A. Chlamydia: Five years A.G. (after genome). Curr. Opin. Microbiol. 2004, 7, 85–92. [Google Scholar]
- Vandahl, B.B.; Birkelund, S.; Christiansen, G. Genome and proteome analysis of Chlamydia. Proteomics 2004, 4, 2831–2842. [Google Scholar]
- Angiuoli, S.V.; Hotopp, J.C.; Salzberg, S.L.; Tettelin, H. Improving pan-genome annotation using whole genome multiple alignment. BMC Bioinformatics 2011, 12, 272. [Google Scholar]
- Donati, C.; Hiller, N.L.; Tettelin, H.; Muzzi, A.; Croucher, N.J.; Angiuoli, S.V.; Oggioni, M.; Dunning Hotopp, J.C.; Hu, F.Z.; Riley, D.R.; et al. Structure and dynamics of the pan-genome of Streptococcus pneumoniae and closely related species. Genome Biol. 2010, 11, R107. [Google Scholar] [CrossRef]
- Jacobsen, A.; Hendriksen, R.S.; Aaresturp, F.M.; Ussery, D.W.; Friis, C. The Salmonella enterica Pan-genome. Microb. Ecol. 2011, 62, 487–504. [Google Scholar]
- Iliopoulos, I.; Tsoka, S.; Andrade, M.A.; Enright, A.J.; Carroll, M.; Poullet, P.; Promponas, V.; Liakopoulos, T.; Palaios, G.; Pasquier, C.; et al. Evaluation of annotation strategies using an entire genome sequence. Bioinformatics 2003, 19, 717–726. [Google Scholar] [CrossRef]
- Ouzounis, C.A.; Karp, P.D. The past, present and future of genome-wide re-annotation. Genome Biol. 2002, 3, comment2001.1–comment2001.6. [Google Scholar]
- Collingro, A.; Tischler, P.; Weinmaier, T.; Penz, T.; Heinz, E.; Brunham, R.C.; Read, T.D.; Bavoil, P.M.; Sachse, K.; Kahane, S.; et al. Unity in variety—The pan-genome of the Chlamydiae. Mol. Biol. Evol. 2011, 28, 3253–3270. [Google Scholar] [CrossRef]
- Bertelli, C.; Collyn, F.; Croxatto, A.; Ruckert, C.; Polkinghorne, A.; Kebbi-Beghdadi, C.; Goesmann, A.; Vaughan, L.; Greub, G. The Waddlia genome: A window into chlamydial biology. PLoS One 2010, 5, e10890. [Google Scholar]
- Laing, C.; Buchanan, C.; Taboada, E.N.; Zhang, Y.; Kropinski, A.; Villegas, A.; Thomas, J.E.; Gannon, V.P. Pan-genome sequence analysis using Panseq: An online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinformatics 2010, 11, 461. [Google Scholar]
- Lapierre, P.; Gogarten, J.P. Estimating the size of the bacterial pan-genome. Trends Genet. 2009, 25, 107–110. [Google Scholar]
- Janssen, P.; Enright, A.J.; Audit, B.; Cases, I.; Goldovsky, L.; Harte, N.; Kunin, V.; Ouzounis, C.A. COmplete GENome Tracking (COGENT): A flexible data environment for computational genomics. Bioinformatics 2003, 19, 1451–1452. [Google Scholar]
- Goldovsky, L.; Janssen, P.; Ahren, D.; Audit, B.; Cases, I.; Darzentas, N.; Enright, A.J.; Lopez-Bigas, N.; Peregrin-Alvarez, J.M.; Smith, M.; et al. CoGenT++: An extensive and extensible data environment for computational genomics. Bioinformatics 2005, 21, 3806–3810. [Google Scholar]
- Promponas, V.J.; Enright, A.J.; Tsoka, S.; Kreil, D.P.; Leroy, C.; Hamodrakas, S.; Sander, C.; Ouzounis, C.A. CAST: An iterative algorithm for the complexity analysis of sequence tracts. Complexity analysis of sequence tracts. Bioinformatics 2000, 16, 915–922. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Enright, A.J.; van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar]
- Lefebure, T.; Bitar, P.D.; Suzuki, H.; Stanhope, M.J. Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol. Evol. 2010, 2, 646–655. [Google Scholar]
- Smith, M.; Kunin, V.; Goldovsky, L.; Enright, A.J.; Ouzounis, C.A. MagicMatch—Cross-referencing sequence identifiers across databases. Bioinformatics 2005, 21, 3429–3430. [Google Scholar]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2011, 39, D38–D51. [Google Scholar]
- Pellegrini, M.; Marcotte, E.M.; Thompson, M.J.; Eisenberg, D.; Yeates, T.O. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. USA 1999, 96, 4285–4288. [Google Scholar]
- Kunin, V.; Ahren, D.; Goldovsky, L.; Janssen, P.; Ouzounis, C.A. Measuring genome conservation across taxa: Divided strains and united kingdoms. Nucleic Acids Res. 2005, 33, 616–621. [Google Scholar]
- Snel, B.; Huynen, M.A.; Dutilh, B.E. Genome trees and the nature of genome evolution. Annu. Rev. Microbiol. 2005, 59, 191–209. [Google Scholar]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar]
- The 10 Supplements can be accessed at http://dx.doi.org/10.5061/dryad.rr064j8q/.
- Heinz, E.; Tischler, P.; Rattei, T.; Myers, G.; Wagner, M.; Horn, M. Comprehensive in silico prediction and analysis of chlamydial outer membrane proteins reflects evolution and life style of the Chlamydiae. BMC Genomics 2009, 10, 634. [Google Scholar]
- Stone, C.B.; Bulir, D.C.; Gilchrist, J.D.; Toor, R.K.; Mahony, J.B. Interactions between flagellar and type III secretion proteins in Chlamydia pneumoniae. BMC Microbiol. 2010, 10, 18. [Google Scholar]
- Muschiol, S.; Boncompain, G.; Vromman, F.; Dehoux, P.; Normark, S.; Henriques-Normark, B.; Subtil, A. Identification of a family of effectors secreted by the type III secretion system that are conserved in pathogenic Chlamydiae. Infect. Immun. 2011, 79, 571–580. [Google Scholar]
- Ouzounis, C.; Bork, P.; Sander, C. The modular structure of NifU proteins. Trends Biochem. Sci. 1994, 19, 199–200. [Google Scholar]
- Ouzounis, C.; Sander, C. Homology of the NifS family of proteins to a new class of pyridoxal phosphate-dependent enzymes. FEBS Lett. 1993, 322, 159–164. [Google Scholar]
- Devos, D.P.; Reynaud, E.G. Evolution. Intermediate steps. Science 2010, 330, 1187–1188. [Google Scholar] [CrossRef]
- McInerney, J.O.; Martin, W.F.; Koonin, E.V.; Allen, J.F.; Galperin, M.Y.; Lane, N.; Archibald, J.M.; Embley, T.M. Planctomycetes and eukaryotes: A case of analogy not homology. Bioessays 2011, 33, 810–817. [Google Scholar]
- Kunin, V.; Ouzounis, C.A. GeneTRACE-reconstruction of gene content of ancestral species. Bioinformatics 2003, 19, 1412–1416. [Google Scholar]
- Bush, R.M.; Everett, K.D. Molecular evolution of the Chlamydiaceae. Int. J. Syst. Evol. Microbiol. 2001, 51, 203–220. [Google Scholar]
- Janssen, P.J.; Audit, B.; Ouzounis, C.A. Strain-specific genes of Helicobacter pylori: Distribution, function and dynamics. Nucleic Acids Res. 2001, 29, 4395–4404. [Google Scholar]
- Brinkman, F.S.; Blanchard, J.L.; Cherkasov, A.; Av-Gay, Y.; Brunham, R.C.; Fernandez, R.C.; Finlay, B.B.; Otto, S.P.; Ouellette, B.F.; Keeling, P.J.; et al. Evidence that plant-like genes in Chlamydia species reflect an ancestral relationship between Chlamydiaceae, cyanobacteria, and the chloroplast. Genome Res. 2002, 12, 1159–1167. [Google Scholar] [CrossRef]
- Huang, J.; Gogarten, J.P. Did an ancient chlamydial endosymbiosis facilitate the establishment of primary plastids? Genome Biol. 2007, 8, R99. [Google Scholar] [CrossRef]
- Becker, B.; Hoef-Emden, K.; Melkonian, M. Chlamydial genes shed light on the evolution of photoautotrophic eukaryotes. BMC Evol. Biol. 2008, 8, 203. [Google Scholar]
- Moustafa, A.; Reyes-Prieto, A.; Bhattacharya, D. Chlamydiae has contributed at least 55 genes to Plantae with predominantly plastid functions. PLoS One 2008, 3, e2205. [Google Scholar]
- Kamneva, O.K.; Liberles, D.A.; Ward, N.L. Genome-wide influence of indel substitutions on evolution of bacteria of the PVC superphylum, revealed using a novel computational method. Genome Biol. Evol. 2010, 2, 870–886. [Google Scholar]
- Moran, N.A.; McCutcheon, J.P.; Nakabachi, A. Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 2008, 42, 165–190. [Google Scholar]
- Merhej, V.; Royer-Carenzi, M.; Pontarotti, P.; Raoult, D. Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol. Direct 2009, 4, 13. [Google Scholar]
- Harris, S.R.; Clarke, I.N.; Seth-Smith, H.M.; Solomon, A.W.; Cutcliffe, L.T.; Marsh, P.; Skilton, R.J.; Holland, M.J.; Mabey, D.; Peeling, R.W.; et al. Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat. Genet. 2012, 44, 413–419. [Google Scholar]
Appendix
Appendix-Table 1.
Core gene and protein families in the Chlamydiales.
| Cluster ID | Lead Sequence | Master Sequence | Function Annotation from Master Sequence |
|---|---|---|---|
| 181 | CABO-LLG-01-000000 | CTRA-DUW-01-000647 | NA |
| 182 | CABO-LLG-01-000003 | CTRA-DUW-01-000644 | ENZYME UDP-N-acetylglucosamine pyrophosphorylase [EC] 2.7.7.23 |
| 183 | CABO-LLG-01-000004 | CTRA-DLC-01-000248 | FUNCTION PhoB-like protein |
| 184 | CABO-LLG-01-000015 | CTRA-DLC-01-000228 | FUNCTION RecA protein |
| 185 | PACA-UV7-01-001616 | CTRA-DUW-01-000487 | NA |
| 186 | CABO-LLG-01-000023 | CTRA-DUW-01-000658 | NA |
| 187 | CABO-LLG-01-000024 | CTRA-DUW-01-000624 | FUNCTION RNA Polymerase Sigma-54 factor RpoN |
| 188 | CABO-LLG-01-000026 | CTRA-DUW-01-000622 | ENZYME Uracil DNA glycosylase [EC] 3.2.2.- |
| 189 | CABO-LLG-01-000028 | CTRA-DUW-01-000620 | SIMILAR-TO NTPase HAM1 homolog [EC] 3.6.1.15 |
| 190 | CABO-LLG-01-000029 | CTRA-DLC-01-000273 | NA |
| 191 | CABO-LLG-01-000032 | CTRA-DUW-01-000616 | NA |
| 192 | CABO-LLG-01-000034 | CTRA-DLC-01-000278 | FUNCTION Peptidoglycan-associated lipoprotein |
| 193 | CABO-LLG-01-000035 | CTRA-DLC-01-000279 | FUNCTION TolB macromolecule uptake homolog |
| 194 | CABO-LLG-01-000037 | CTRA-DUW-01-000611 | FUNCTION TolR/ExbD macromolecule uptake homolog |
| 195 | CABO-LLG-01-000040 | CTRA-DLC-01-000284 | FUNCTION protein translocase TatD/MttC homolog |
| 196 | CABO-LLG-01-000047 | CTRA-DUW-01-000600 | ENZYME enolase [EC] 4.2.1.11 |
| 197 | CABO-LLG-01-000048 | CTRA-DUW-01-000599 | FUNCTION Excinuclease ABC subunit B |
| 198 | CABO-LLG-01-000049 | CTRA-DUW-01-000598 | ENZYME Tryptophanyl-tRNA Synthetase [EC] 6.1.1.2 |
| 199 | CTRA-G22-01-000161 | CTRA-DUW-01-000746 | ENZYME Seryl-tRNA Synthetase [EC] 6.1.1.11 |
| 200 | CABO-LLG-01-000054 | CTRA-DUW-01-000593 | FUNCTION Nickel transporter CnrT homolog |
| 201 | CABO-LLG-01-000061 | CTRA-DUW-01-000586 | NA |
| 202 | CABO-LLG-01-000062 | CTRA-DUW-01-000585 | FUNCTION type II secretion system protein D homolog |
| 203 | CABO-LLG-01-000063 | CTRA-DUW-01-000584 | FUNCTION type II secretion system protein E homolog |
| 204 | CABO-LLG-01-000064 | CTRA-DLC-01-000308 | FUNCTION type II secretion system protein F homolog |
| 205 | CABO-LLG-01-000065 | CTRA-DLC-01-000309 | NA |
| 206 | CABO-LLG-01-000070 | CTRA-DUW-01-000577 | FUNCTION protein secretion system YscT homolog |
| 207 | CABO-LLG-01-000072 | CTRA-DLC-01-000316 | FUNCTION protein secretion system YscR homolog |
| 208 | CABO-LLG-01-000073 | CTRA-DEC-01-000317 | FUNCTION protein secretion system YscL homolog |
| 209 | CABO-LLG-01-000074 | CTRA-DUW-01-000573 | NA |
| 210 | CABO-LLG-01-000076 | CTRA-DUW-01-000571 | ENZYME lipoate synthase [EC] 2.8.1.- |
| 211 | CABO-LLG-01-000081 | CTRA-DUW-01-000714 | ENZYME Endonuclease III [EC] 4.2.99.18 |
| 212 | CABO-LLG-01-000083 | CTRA-DUW-01-000716 | ENZYME Phosphatidylserine decarboxylase [EC] 4.1.1.65 |
| 213 | CABO-LLG-01-000085 | CTRA-DUW-01-000718 | FUNCTION preprotein translocase subunit SecA |
| 214 | CABO-LLG-01-000089 | CTRA-DUW-01-000722 | ENZYME ATP-dependent Clp protease ATP-binding subunit ClpX [EC] |
| 215 | CABO-LLG-01-000091 | CTRA-DUW-01-000724 | FUNCTION Trigger factor |
| 216 | CABO-LLG-01-000093 | CTRA-DUW-01-000726 | FUNCTION Rod shape-determining protein MreB |
| 217 | CABO-LLG-01-000094 | CTRA-DUW-01-000727 | ENZYME Phosphoenolpyruvate carboxykinase (GTP) [EC] 4.1.1.32 |
| 218 | CABO-LLG-01-000098 | CTRA-DUW-01-000731 | ENZYME Glycerol-3-phosphate dehydrogenase [NAD+] [EC] 1.1.1.8 |
| 219 | CABO-LLG-01-000099 | CTRA-DUW-01-000732 | ENZYME UDP-N-acetylhexosamine pyrophosphorylase [EC] 2.7.7.- |
| 220 | CCAV-GPI-01-000128 | CTRA-DUW-01-000503 | FUNCTION Transcription termination factor Rho |
| 221 | CABO-LLG-01-000104 | CTRA-DUW-01-000737 | DOMAIN NifU |
| 222 | PACA-HAL-01-002518 | CTRA-DUW-01-000261 | ENZYME NifS aminotransferase [EC] -.-.-.- |
| 223 | CABO-LLG-01-000109 | CTRA-DUW-01-000742 | ENZYME Biotin-[acetyl-CoA-carboxylase] synthetase [EC] 6.3.4.15 |
| 224 * | CABO-LLG-01-000121 | CTRA-DUW-01-000754 | DOMAIN SET |
| 225 | CABO-LLG-01-000122 | CTRA-DUW-01-000755 | SIMILAR-TO metallo-beta-lactamase [EC] 3.5.-.- |
| 226 | CABO-LLG-01-000123 | CTRA-DUW-01-000756 | FUNCTION Cell division protein FtsK C-terminus |
| 227 | CABO-LLG-01-000125 | CTRA-DUW-01-000757 | NA |
| 228 | CABO-LLG-01-000126 | CTRA-DUW-01-000758 | FUNCTION preprotein translocase complex subunit YajC |
| 229 | CABO-LLG-01-000130 | CTRA-DUW-01-000762 | ENZYME Protoporphyrinogen oxidase HemY [EC] 1.3.3.4 |
| 230 | CABO-LLG-01-000132 | CTRA-DUW-01-000764 | ENZYME Uroporphyrinogen decarboxylase HemE [EC] 4.1.1.37 |
| 231 | CABO-LLG-01-000134 | CTRA-DLC-01-000129 | ENZYME Alanyl-tRNA Synthetase [EC] 6.1.1.7 |
| 232 | CABO-LLG-01-000135 | CTRA-DUW-01-000767 | ENZYME Transketolase [EC] 2.2.1.1 |
| 233 | CABO-LLG-01-000136 | CTRA-DUW-01-000768 | SIMILAR-TO AMP nucleosidase [EC] 3.2.2.4 |
| 234 | CABO-LLG-01-000142 | CTRA-DUW-01-000774 | ENZYME Phospho-N-acetylmuramoyl-pentapeptide-transferase [EC] |
| 235 | CABO-LLG-01-000143 | CTRA-DUW-01-000775 | ENZYME UDP-N-acetylmuramoylalanine-D-glutamate ligase [EC] |
| 236 | CABO-LLG-01-000144 | CTRA-DUW-01-000776 | SIMILAR-TO N-acetylmuramoyl-L-alanine amidase C-terminus [EC] |
| 237 | CABO-LLG-01-000146 | CTRA-DLC-01-000117 | ENZYME UDP-N-acetylglucosamine-N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase |
| 238 | CABO-S26-01-000517 | CTRA-DUW-01-000125 | ENZYME Biotin carboxylase [EC] 6.3.4.14 |
| 239 | CABO-LLG-01-000150 | CTRA-DUW-01-000781 | NA |
| 240 | CABO-LLG-01-000155 | CTRA-DUW-01-000786 | NA |
| 241 | CABO-LLG-01-000157 | CTRA-DUW-01-000788 | ENZYME bis(5'-nucleosyl)-tetraphosphatase [EC] 3.6.1.17 |
| 242 | CABO-LLG-01-000168 | CTRA-DLC-01-000098 | ENZYME Cysteinyl-tRNA Synthetase [EC] 6.1.1.16 |
| 243 | CABO-LLG-01-000173 | CTRA-DUW-01-000804 | FUNCTION Ribosomal protein S14 |
| 244 | CABO-LLG-01-000174 | CTRA-DUW-01-000805 | NA |
| 245 | CABO-LLG-01-000176 | CTRA-DUW-01-000808 | ENZYME Excinuclease ABC subunit C [EC] -.-.-.- |
| 246 | CABO-LLG-01-000177 | CTRA-DUW-01-000809 | FUNCTION DNA mismatch repair protein MutS |
| 247 | CABO-LLG-01-000184 | CTRA-DUW-01-000815 | ENZYME CDP-diacylglycerol-glycerol-3-phosphate |
| 248 | CABO-LLG-01-000185 | CTRA-DUW-01-000816 | ENZYME Glycogen synthase [EC] 2.4.1.21 2 |
| 249 | CABO-LLG-01-000186 | CTRA-DUW-01-000817 | FUNCTION Ribosomal protein L25 |
| 250 | CABO-LLG-01-000187 | CTRA-DUW-01-000818 | ENZYME Peptidyl-tRNA hydrolase [EC] 3.1.1.29 |
| 251 | CABO-LLG-01-000188 | CTRA-DUW-01-000819 | FUNCTION Ribosomal protein S6 |
| 252 | CABO-LLG-01-000189 | CTRA-DUW-01-000820 | FUNCTION Ribosomal protein S18 |
| 253 | CABO-LLG-01-000190 | CTRA-DUW-01-000821 | FUNCTION Ribosomal protein L9 |
| 254 * | CABO-LLG-01-000193 | CTRA-DUW-01-000823 | NA |
| 255 * | CABO-LLG-01-000194 | CTRA-DUW-01-000824 | SIMILAR-TO Small-peptide endopeptidase [EC] 3.4.24.55 |
| 256 | CABO-LLG-01-000195 | CTRA-DLC-01-000073 | ENZYME Glycerol-3-phosphate acyltransferase [EC] 2.3.1.15 |
| 257 | CABO-LLG-01-000196 | CTRA-DLC-01-000072 | ENZYME Ribonuclease E [EC] 3.1.4.- |
| 258 | CABO-LLG-01-000197 | CTRA-DLC-01-000071 | NA |
| 259 | CABO-LLG-01-000214 | CTRA-DLC-01-000063 | ENZYME Glucosamine-fructose-6-phosphate aminotransferase [EC] |
| 260 | CABO-LLG-01-000218 | CTRA-DUW-01-000840 | ENZYME Succinyl-CoA synthetase beta chain [EC] 6.2.1.5 |
| 261 | CABO-LLG-01-000222 | CTRA-DUW-01-000843 | SIMILAR-TO Small-peptide endopeptidase [EC] 3.4.24.55 |
| 262 | CABO-LLG-01-000224 | CTRA-DUW-01-000845 | ENZYME CDP-diacylglycerol-serine O-phosphatidyltransferase [EC] |
| 263 | CABO-LLG-01-000229 | CTRA-DUW-01-000850 | ENZYME UDP-N-acetylenolpyruvoylglucosamine reductase [EC] |
| 264 | CABO-LLG-01-000230 | CTRA-DLC-01-000047 | FUNCTION Transcription termination protein NusB |
| 265 | CABO-LLG-01-000231 | CTRA-DLC-01-000046 | NA |
| 266 | CABO-LLG-01-000233 | CTRA-DUW-01-000854 | FUNCTION Ribosomal protein L20 |
| 267 | CABO-LLG-01-000234 | CTRA-DUW-01-000855 | ENZYME Phenylalanyl-tRNA Synthetase alpha chain [EC] 6.1.1.20 |
| 268 | CABO-LLG-01-000236 | CTRA-DUW-01-000857 | NA |
| 269 | CABO-LLG-01-000237 | CTRA-DUW-01-000858 | NA |
| 270 | CABO-LLG-01-000240 | CTRA-DUW-01-000861 | ENZYME Polynucleotide phosphorylase [EC] 2.7.7.8 |
| 271 | CABO-LLG-01-000241 | CTRA-DUW-01-000862 | NA |
| 272 * | CABO-LLG-01-000254 | CTRA-DUW-01-000874 | FUNCTION ABC transporter, ATP-binding protein N-terminus |
| 273 | CABO-LLG-01-000267 | CTRA-DUW-01-000385 | ENZYME Glucose-6-phosphate isomerase [EC] 5.3.1.9 |
| 274 | CABO-LLG-01-000269 | CTRA-DLC-01-000502 | ENZYME Malate dehydrogenase [EC] 1.1.1.82 |
| 275 | CABO-LLG-01-000271 | CTRA-DUW-01-000382 | SIMILAR-TO D-Amino Acid Dehydrogenase [EC] 1.-.-.- |
| 276 * | CABO-LLG-01-000276 | CTRA-DLC-01-000508 | ENZYME 3-dehydroquinate dehydratase [EC] 4.2.1.10 |
| 277 | CPRO-UWE-01-000881 | CTRA-DUW-01-000373 | ENZYME 3-phosphoshikimate 1-carboxyvinyltransferase [EC] |
| 278 | CABO-LLG-01-000277 | CTRA-DUW-01-000376 | ENZYME 3-dehydroquinate synthase [EC] 4.6.1.3 |
| 279 | CABO-LLG-01-000278 | CTRA-DUW-01-000375 | ENZYME Chorismate synthase [EC] 4.6.1.4 |
| 280 | CABO-LLG-01-000288 | CTRA-DUW-01-000371 | ENZYME Dihydrodipicolinate reductase [EC] 1.3.1.26 |
| 281 | CABO-LLG-01-000290 | CTRA-DUW-01-000369 | ENZYME Aspartokinase [EC] 2.7.2.4 |
| 282 | CABO-LLG-01-000298 | CTRA-DUW-01-000328 | NA |
| 283 | SNEG-ZXX-01-000625 | CTRA-DLC-01-000783 | FUNCTION Translation initiation factor IF-2 |
| 284 | CABO-LLG-01-000304 | CTRA-DUW-01-000322 | FUNCTION Ribosomal protein L11 |
| 285 | CABO-LLG-01-000305 | CTRA-DUW-01-000321 | FUNCTION Ribosomal protein L1 |
| 286 | CABO-LLG-01-000306 | CTRA-DUW-01-000320 | FUNCTION Ribosomal protein L10 |
| 287 | CABO-LLG-01-000308 | CTRA-DUW-01-000318 | ENZYME DNA-directed RNA polymerase beta subunit [EC] 2.7.7.6 |
| 288 | CABO-LLG-01-000309 | CTRA-DUW-01-000317 | ENZYME DNA-directed RNA polymerase beta prime subunit [EC] |
| 289 | CABO-LLG-01-000312 | CTRA-DUW-01-000314 | NA |
| 290 | CABO-LLG-01-000313 | CTRA-DLC-01-000569 | ENZYME vacuolar ATPase proteolipid subunit E [EC] 3.6.1.34 |
| 291 | CABO-LLG-01-000314 | CTRA-DUW-01-000312 | NA |
| 292 | CABO-LLG-01-000317 | CTRA-DUW-01-000309 | ENZYME vacuolar ATPase proteolipid subunit D [EC] 3.6.1.34 |
| 293 | CABO-LLG-01-000320 | CTRA-DLC-01-000576 | NA |
| 294 | CABO-LLG-01-000324 | CTRA-DUW-01-000337 | ENZYME Pyruvate kinase [EC] 2.7.1.40 |
| 295 | CPRO-UWE-01-001632 | CTRA-DUW-01-000012 | NA |
| 296 | CABO-LLG-01-000328 | CTRA-DUW-01-000013 | ENZYME Cytochrome Oxidase D subunit I [EC] 1.10.3.- |
| 297 | CABO-LLG-01-000329 | CTRA-DUW-01-000014 | ENZYME Cytochrome Oxidase D subunit II [EC] 1.10.3.- |
| 298 | CABO-LLG-01-000331 | CTRA-DLC-01-000860 | NA |
| 299 | CABO-LLG-01-000332 | CTRA-DLC-01-000861 | NA |
| 300 | CABO-LLG-01-000333 | CTRA-DUW-01-000015 | FUNCTION PhoH-like protein |
| 301 | CABO-LLG-01-000337 | CTRA-DLC-01-000856 | NA |
| 302 | CABO-LLG-01-000338 | CTRA-DUW-01-000022 | FUNCTION Ribosomal protein L31 |
| 303 | CABO-LLG-01-000342 | CTRA-DUW-01-000026 | FUNCTION Ribosomal protein S16 |
| 304 | CABO-LLG-01-000343 | CTRA-DUW-01-000027 | ENZYME tRNA (guanine N-1) methyltransferase [EC] 2.1.1.31 |
| 305 | CABO-LLG-01-000344 | CTRA-DUW-01-000028 | FUNCTION Ribosomal protein L19 |
| 306 | CABO-LLG-01-000345 | CTRA-DUW-01-000029 | ENZYME Ribonuclease HII [EC] 3.1.26.4 |
| 307 | CABO-LLG-01-000346 | CTRA-DUW-01-000030 | ENZYME Guanylate kinase [EC] 2.7.4.8 |
| 308 | CABO-LLG-01-000358 | CTRA-DUW-01-000215 | ENZYME Ribose 5-phosphate isomerase A [EC] 5.3.1.6 |
| 309 | CABO-LLG-01-000359 | CTRA-DLC-01-000666 | NA |
| 310 | CABO-LLG-01-000360 | CTRA-DUW-01-000213 | NA |
| 311 | CABO-LLG-01-000368 | CTRA-DLC-01-000765 | NA |
| 312 | CABO-LLG-01-000374 | CTRA-DUW-01-000147 | ENZYME DNA ligase (NAD+) [EC] 6.5.1.2 |
| 313 | CABO-LLG-01-000379 | CTRA-DUW-01-000210 | ENZYME 3-deoxy-d-manno-octulosonic-acid transferase [EC] 2.-.-.- |
| 314 | CABO-LLG-01-000392 | CTRA-DUW-01-000186 | NA |
| 315 | CABO-LLG-01-000393 | CTRA-DUW-01-000185 | ENZYME CTP synthetase [EC] 6.3.4.2 |
| 316 | CABO-LLG-01-000404 | CTRA-DUW-01-000195 | ENZYME Queuine tRNA-ribosyltransferase [EC] 2.4.2.29 |
| 317 | CABO-LLG-01-000420 | CTRA-DUW-01-000132 | NA |
| 318 | CABO-LLG-01-000423 | CTRA-DUW-01-000199 | ENZYME O-sialoglycoprotein endopeptidase [EC] 3.4.24.57 |
| 319 | CABO-LLG-01-000425 | CTRA-DUW-01-000187 | NA |
| 320 | CABO-LLG-01-000426 | CTRA-DUW-01-000188 | ENZYME Glucose-6-phosphate dehydrogenase [EC] -.-.-.- |
| 321 | CABO-LLG-01-000432 | CTRA-DLC-01-000753 | FUNCTION Ribosomal protein S9 |
| 322 | CABO-LLG-01-000433 | CTRA-DUW-01-000126 | FUNCTION Ribosomal protein L13 |
| 323 | CABO-LLG-01-000435 | CTRA-DUW-01-000152 | NA |
| 324 | CABO-LLG-01-000448 | CTRA-DUW-01-000138 | FUNCTION Sua5 homolog |
| 325 | CABO-LLG-01-000451 | CTRA-DUW-01-000190 | ENZYME Thymidylate kinase (dTMP kinase) [EC] 2.7.4.9 |
| 326 | CABO-LLG-01-000459 | CTRA-DUW-01-000217 | ENZYME Fructose-bisphosphate aldolase class I [EC] 4.1.2.13 |
| 327 | CABO-LLG-01-000471 | CTRA-DUW-01-000239 | FUNCTION acyl carrier protein ACP |
| 328 | PACA-UV7-01-000731 | CTRA-DUW-01-000105 | ENZYME Enoyl-[acyl-carrier protein] reductase (NADH) [EC] |
| 329 | CABO-LLG-01-000473 | CTRA-DLC-01-000640 | ENZYME Malonyl CoA-acyl carrier protein transacylase [EC] |
| 330 | CABO-LLG-01-000474 | CTRA-DLC-01-000639 | ENZYME 3-oxoacyl-[acyl-carrier-protein] synthase III [EC] |
| 331 | CABO-LLG-01-000475 | CTRA-DUW-01-000243 | FUNCTION Recombination protein RecR homolog |
| 332 | CABO-LLG-01-000477 | CTRA-DUW-01-000245 | NA |
| 333 | CPNE-TW1-01-000387 | CTRA-DUW-01-000055 | ENZYME 2-oxoglutarate dehydrogenase E1 component [EC] 1.2.4.2 |
| 334 | CABO-LLG-01-000486 | CTRA-DUW-01-000254 | FUNCTION Inner-membrane protein YidC |
| 335 | CABO-LLG-01-000489 | CTRA-DUW-01-000101 | ENZYME holo-[acyl-carrier protein] synthase [EC] 2.7.8.7 |
| 336 | CABO-LLG-01-000490 | CTRA-DUW-01-000100 | ENZYME Thioredoxin reductase (NADPH) [EC] 1.6.4.5 |
| 337 | CABO-LLG-01-000494 | CTRA-DUW-01-000096 | FUNCTION Ribosome-binding factor A RbfA |
| 338 | CABO-LLG-01-000496 | CTRA-DUW-01-000094 | ENZYME Riboflavin kinase [EC] 2.7.1.26 |
| 339 | CABO-LLG-01-000499 | CTRA-DUW-01-000090 | NA |
| 340 | CABO-LLG-01-000500 | CTRA-DUW-01-000089 | NA |
| 341 | CABO-LLG-01-000501 | CTRA-DLC-01-000793 | FUNCTION Ribosomal protein L28 |
| 342 | CABO-LLG-01-000508 | CTRA-DLC-01-000801 | ENZYME Methylenetetrahydrofolate dehydrogenase [EC] 1.5.1.15 |
| 343 | CABO-LLG-01-000509 | CTRA-DUW-01-000078 | FUNCTION Thiamine biosynthesis lipoprotein ApbE precursor |
| 344 | CABO-LLG-01-000510 | CTRA-DUW-01-000077 | FUNCTION Small protein B SmpB homolog |
| 345 | CABO-LLG-01-000511 | CTRA-DUW-01-000076 | ENZYME DNA polymerase III beta chain [EC] 2.7.7.7 |
| 346 | CABO-LLG-01-000514 | CTRA-DUW-01-000073 | SIMILAR-TO zinc protease [EC] -.-.-.- |
| 347 | CABO-LLG-01-000516 | CTRA-DUW-01-000071 | FUNCTION ABC transporter, permease protein TroD |
| 348 | CABO-LLG-01-000519 | CTRA-DUW-01-000068 | FUNCTION periplasmic substrate binding protein TroA |
| 349 | CPSI-CAL-01-000799 | CTRA-DUW-01-000423 | FUNCTION high-affinity ZnuA homolog |
| 350 | CABO-LLG-01-000520 | CTRA-DLC-01-000812 | NA |
| 351 | CABO-LLG-01-000523 | CTRA-DUW-01-000064 | ENZYME 6-phosphogluconate dehydrogenase [EC] 1.1.1.44 |
| 352 | CABO-LLG-01-000524 | CTRA-DUW-01-000063 | ENZYME Tyrosyl-tRNA Synthetase [EC] 6.1.1.1 |
| 353 | CABO-LLG-01-000535 | CTRA-DLC-01-000825 | NA |
| 354 | CABO-LLG-01-000541 | CTRA-DLC-01-000831 | NA |
| 355 | CABO-LLG-01-000544 | CTRA-DUW-01-000045 | FUNCTION single-stranded DNA-binding protein SSB |
| 356 | CABO-LLG-01-000545 | CTRA-DUW-01-000044 | NA |
| 357 | CABO-LLG-01-000547 | CTRA-DUW-01-000042 | NA |
| 358 | CABO-LLG-01-000554 | CTRA-DLC-01-000619 | ENZYME Protein phosphatase 2C [EC] 3.1.3.16 |
| 359 | CABO-LLG-01-000558 | CTRA-DUW-01-000257 | NA |
| 360 | CABO-LLG-01-000560 | CTRA-DUW-01-000108 | ENZYME A/G-specific adenine glycosylase [EC] 3.2.2.- |
| 361 | CABO-LLG-01-000564 | CTRA-DUW-01-000104 | NA |
| 362 | CABO-LLG-01-000571 | CTRA-DUW-01-000268 | ENZYME Acetyl-coenzyme A carboxylase carboxyl transferase |
| 363 | CABO-LLG-01-000574 | CTRA-DUW-01-000271 | ENZYME N-acetylmuramoyl-L-alanine amidase AmiB [EC] 3.5.1.28 |
| 364 | CABO-LLG-01-000577 | CTRA-DUW-01-000273 | FUNCTION Penicillin-binding protein 3 |
| 365 | CABO-LLG-01-000578 | CTRA-DUW-01-000274 | NA |
| 366 | CABO-LLG-01-000581 | CTRA-DUW-01-000277 | DOMAIN TPR |
| 367 | CABO-LLG-01-000585 | CTRA-DLC-01-000601 | NA |
| 368 | CABO-LLG-01-000586 | CTRA-DUW-01-000282 | NA |
| 369 | CABO-LLG-01-000587 | CTRA-DLC-01-000599 | NA |
| 370 | CPEC-E58-01-000614 | CTRA-DUW-01-000284 | NA |
| 371 | CABO-LLG-01-000591 | CTRA-DLC-01-000597 | FUNCTION Glycine cleavage system H protein |
| 372 | CABO-LLG-01-000594 | CTRA-DUW-01-000288 | SIMILAR-TO Lipoate-protein ligase A [EC] 6.3.4.- |
| 373 | CABO-LLG-01-000596 | CTRA-DUW-01-000290 | ENZYME tRNA (5-methylaminomethyl-2-thiouridylate)-methyltransferase |
| 374 | CABO-LLG-01-000601 | CTRA-DUW-01-000293 | ENZYME Nitrogen regulatory IIA protein A component [EC] 2.7.1.69 |
| 375 | WCHO-WSU-01-000243 | CTRA-DUW-01-000294 | ENZYME Nitrogen regulatory IIA protein A component [EC] 2.7.1.69 |
| 376 | CABO-LLG-01-000603 | CTRA-DUW-01-000295 | ENZYME dUTP pyrophosphatase [EC] 3.6.1.23 |
| 377 | CABO-LLG-01-000608 | CTRA-DUW-01-000300 | ENZYME Ribonuclease III [EC] 3.1.26.3 |
| 378 | CABO-LLG-01-000609 | CTRA-DLC-01-000581 | FUNCTION DNA repair protein RadA |
| 379 | CABO-LLG-01-000610 | CTRA-DUW-01-000302 | ENZYME Porphobilinogen deaminase [EC] 4.3.1.8 |
| 380 | CABO-LLG-01-000616 | CTRA-DUW-01-000340 | NA |
| 381 | CABO-LLG-01-000623 | CTRA-DUW-01-000346 | DOMAIN DnaJ |
| 382 | CABO-LLG-01-000624 | CTRA-DLC-01-000536 | FUNCTION Ribosomal protein S21 |
| 383 | CABO-LLG-01-000628 | CTRA-DUW-01-000351 | ENZYME Aryl-sulfate sulphohydrolase [EC] 3.1.6.1 |
| 384 | CABO-LLG-01-000631 | CTRA-DUW-01-000354 | FUNCTION Septum formation protein Maf homolog |
| 385 | CABO-LLG-01-000632 | CTRA-DUW-01-000355 | NA |
| 386 | WCHO-WSU-01-000567 | CTRA-DUW-01-000392 | NA |
| 387 | CABO-LLG-01-000633 | CTRA-DUW-01-000356 | NA |
| 388 | CABO-LLG-01-000636 | CTRA-DUW-01-000333 | ENZYME Triosephosphate isomerase [EC] 5.3.1.1 |
| 389 | CABO-LLG-01-000637 | CTRA-DUW-01-000334 | ENZYME Exonuclease VII large subunit [EC] 3.1.11.6 |
| 390 | CABO-LLG-01-000641 | CTRA-DUW-01-000360 | ENZYME Dimethyladenosine transferase [EC] 2.1.1.- |
| 391 | CABO-LLG-01-000642 | CTRA-DUW-01-000361 | NA |
| 392 | CABO-LLG-01-000643 | CTRA-DUW-01-000362 | DOMAIN Thioredoxin |
| 393 | CABO-LLG-01-000646 | CTRA-DLC-01-000868 | NA |
| 394 | CABO-LLG-01-000647 | CTRA-DLC-01-000869 | ENZYME Ribonuclease HII [EC] 3.1.26.4 |
| 395 | CABO-LLG-01-000651 | CTRA-DUW-01-000004 | ENZYME glutamyl-tRNA (Gln) amidotransferase, subunit B [EC] |
| 396 | CABO-LLG-01-000670 | CTRA-DUW-01-000386 | NA |
| 397 | CABO-LLG-01-000671 | CTRA-DUW-01-000387 | SIMILAR-TO metallo-beta-lactamase [EC] 3.5.-.- |
| 398 | CABO-LLG-01-000684 | CTRA-DLC-01-000488 | NA |
| 399 | CABO-LLG-01-000686 | CTRA-DUW-01-000396 | NA |
| 400 | CABO-LLG-01-000690 | CTRA-DLC-01-000484 | FUNCTION Heat-inducible transcription repressor HrcA |
| 401 | CABO-LLG-01-000691 | CTRA-DUW-01-000403 | FUNCTION GrpE protein |
| 402 | CABO-LLG-01-000698 | CTRA-DUW-01-000432 | NA |
| 403 | CABO-LLG-01-000701 | CTRA-DUW-01-000435 | NA |
| 404 | CABO-LLG-01-000705 | CTRA-DUW-01-000438 | ENZYME ubiquinone/menaquinone biosynthesis methlytransferase |
| 405 | CABO-LLG-01-000706 | CTRA-DLC-01-000449 | NA |
| 406 | CABO-LLG-01-000707 | CTRA-DUW-01-000440 | ENZYME Diaminopimelate epimerase [EC] 5.1.1.7 |
| 407 | CABO-LLG-01-000709 | CTRA-DLC-01-000446 | ENZYME Serine hydroxymethyltransferase [EC] 2.1.2.1 |
| 408 | CABO-LLG-01-000713 | CTRA-DUW-01-000406 | NA |
| 409 | CABO-LLG-01-000714 | CTRA-DLC-01-000479 | NA |
| 410 | CABO-LLG-01-000717 | CTRA-DUW-01-000410 | ENZYME Lipid A 4'-kinase [EC] 2.7.1.130 |
| 411 | CABO-LLG-01-000722 | CTRA-DLC-01-000471 | FUNCTION DnaK suppressor protein |
| 412 | CABO-LLG-01-000723 | CTRA-DUW-01-000416 | ENZYME Lipoprotein signal peptidase [EC] 3.4.23.36 |
| 413 | CABO-LLG-01-000735 | CTRA-DUW-01-000427 | FUNCTION Ribosomal protein L27 |
| 414 | CABO-LLG-01-000736 | CTRA-DLC-01-000459 | FUNCTION Ribosomal protein L21 |
| 415 | CABO-LLG-01-000738 | CTRA-DUW-01-000444 | NA |
| 416 | CABO-LLG-01-000739 | CTRA-DUW-01-000445 | ENZYME Sulfite reductase (NADPH) flavoprotein alpha-component |
| 417 | CABO-LLG-01-000740 | CTRA-DLC-01-000442 | FUNCTION Ribosomal protein S10 |
| 418 | CABO-LLG-01-000751 | CTRA-DUW-01-000456 | ENZYME Glutamyl-tRNA Synthetase [EC] 6.1.1.17 |
| 419 | CABO-LLG-01-000752 | CTRA-DLC-01-000431 | NA |
| 420 * | CABO-LLG-01-000753 | NA | |
| 421 | CABO-LLG-01-000754 | CTRA-DUW-01-000458 | ENZYME Single-stranded-DNA-specific exonuclease RecJ [EC] |
| 422 | CABO-LLG-01-000759 | CTRA-DUW-01-000463 | ENZYME Cytidylate kinase [EC] 2.7.4.14 |
| 423 | CABO-LLG-01-000761 | CTRA-DUW-01-000465 | ENZYME Arginyl-tRNA Synthetase [EC] 6.1.1.19 |
| 424 | CABO-LLG-01-000762 | CTRA-DUW-01-000466 | ENZYME UDP-N-acetylglucosamine 1-carboxyvinyltransferase [EC] |
| 425 | CABO-LLG-01-000764 | CTRA-DUW-01-000468 | NA |
| 426 | CABO-LLG-01-000778 | CTRA-DUW-01-000480 | NA |
| 427 | CABO-LLG-01-000779 | CTRA-DUW-01-000481 | NA |
| 428 | CABO-LLG-01-000784 | CTRA-DUW-01-000486 | ENZYME Phenylalanyl-tRNA Synthetase beta chain [EC] 6.1.1.20 |
| 429 * | CABO-LLG-01-000789 | CTRA-DUW-01-000491 | FUNCTION Dipeptide binding protein DppA |
| 430 | CABO-LLG-01-000792 | CTRA-DUW-01-000496 | NA |
| 431 | CABO-LLG-01-000793 | CTRA-DUW-01-000497 | ENZYME Protoheme ferro-lyase [EC] 4.99.1.1 |
| 432 | CABO-LLG-01-000794 | CTRA-DUW-01-000498 | FUNCTION Aminoacid-binding periplasmic protein precursor |
| 433 | CABO-LLG-01-000795 | CTRA-DUW-01-000499 | ENZYME HemK modification methylase homolog [EC] -.-.-.- |
| 434 | CABO-LLG-01-000796 | CTRA-DUW-01-000500 | NA |
| 435 | CABO-LLG-01-000801 | CTRA-DLC-01-000386 | DOMAIN ATP-binding |
| 436 | CABO-LLG-01-000802 | CTRA-DUW-01-000505 | ENZYME DNA polymerase I [EC] 2.7.7.7 |
| 437 | CABO-LLG-01-000803 | CTRA-DLC-01-000384 | NA |
| 438 | CABO-LLG-01-000805 | CTRA-DUW-01-000508 | ENZYME CDP-diacylglycerol-glycerol-3-phosphate |
| 439 | CABO-LLG-01-000807 | CTRA-DUW-01-000511 | FUNCTION Glucose inhibited division protein A GidA |
| 440 | CABO-LLG-01-000808 | CTRA-DUW-01-000512 | ENZYME Lipoate-protein ligase A [EC] 6.3.4.- |
| 441 | CABO-LLG-01-000810 | CTRA-DUW-01-000514 | ENZYME Holliday Junction DNA Helicase RuvA [EC] -.-.-.- |
| 442 | CABO-LLG-01-000811 | CTRA-DUW-01-000515 | ENZYME Holliday Junction DNA Helicase RuvC [EC] 3.1.22.4 |
| 443 | CABO-LLG-01-000813 | CTRA-DUW-01-000517 | NA |
| 444 | CABO-LLG-01-000814 | CTRA-DUW-01-000518 | ENZYME Glyceraldehyde 3-phosphate dehydrogenase [EC] 1.2.1.12 |
| 445 | CABO-LLG-01-000820 | CTRA-DUW-01-000524 | FUNCTION Ribosomal protein L15 |
| 446 | CABO-LLG-01-000821 | CTRA-DUW-01-000525 | FUNCTION Ribosomal protein S5 |
| 447 | CABO-LLG-01-000822 | CTRA-DUW-01-000526 | FUNCTION Ribosomal protein L18 |
| 448 | CABO-LLG-01-000824 | CTRA-DUW-01-000528 | FUNCTION Ribosomal protein S8 |
| 449 | CABO-LLG-01-000825 | CTRA-DUW-01-000529 | FUNCTION Ribosomal protein L5 |
| 450 | CABO-LLG-01-000826 | CTRA-DUW-01-000530 | FUNCTION Ribosomal protein L24 |
| 451 | CABO-LLG-01-000827 | CTRA-DUW-01-000531 | FUNCTION Ribosomal protein L14 |
| 452 | CABO-LLG-01-000828 | CTRA-DUW-01-000532 | FUNCTION Ribosomal protein S17 |
| 453 | CABO-LLG-01-000830 | CTRA-DUW-01-000534 | FUNCTION Ribosomal protein L16 |
| 454 | CABO-LLG-01-000831 | CTRA-DUW-01-000535 | FUNCTION Ribosomal protein S3 |
| 455 | CABO-LLG-01-000833 | CTRA-DUW-01-000537 | FUNCTION Ribosomal protein S19 |
| 456 | CABO-LLG-01-000834 | CTRA-DUW-01-000538 | FUNCTION Ribosomal protein L2 |
| 457 | CABO-LLG-01-000835 | CTRA-DUW-01-000539 | FUNCTION Ribosomal protein L23 |
| 458 | CABO-LLG-01-000836 | CTRA-DLC-01-000351 | FUNCTION Ribosomal protein L4 |
| 459 | CABO-LLG-01-000837 | CTRA-DUW-01-000541 | FUNCTION Ribosomal protein L3 |
| 460 * | CABO-LLG-01-000839 | CTRA-DUW-01-000543 | ENZYME Methionyl-tRNA formyltransferase [EC] 2.1.2.9 |
| 461 | CABO-LLG-01-000841 | CTRA-DUW-01-000545 | ENZYME (3R)-hydroxymyristoyl-[acyl carrier protein] dehydratase |
| 462 | CABO-LLG-01-000842 | CTRA-DLC-01-000345 | ENZYME UDP-3-O-[3-hydroxymyristoyl] N-acetylglucosamine |
| 463 | CABO-LLG-01-000843 | CTRA-DUW-01-000547 | ENZYME apolipoprotein N-acyltransferase [EC] 2.3.1. |
| 464 | CABO-LLG-01-000846 | CTRA-DUW-01-000550 | DOMAIN ATP-binding |
| 465 | CABO-LLG-01-000847 | CTRA-DUW-01-000551 | NA |
| 466 | CABO-LLG-01-000849 | CTRA-DUW-01-000553 | ENZYME rRNA methyltransferase SpoU homolog [EC] -.-.-.- |
| 467 | CABO-LLG-01-000852 | CTRA-DUW-01-000556 | ENZYME Histidyl-tRNA Synthetase [EC] 6.1.1.21 |
| 468 | CABO-LLG-01-000855 | CTRA-DUW-01-000558 | ENZYME DNA polymerase III alpha chain [EC] 2.7.7.7 |
| 469 | CABO-LLG-01-000856 | CTRA-DUW-01-000559 | NA |
| 470 | CABO-LLG-01-000857 | CTRA-DLC-01-000331 | NA |
| 471 | CABO-LLG-01-000858 | CTRA-DUW-01-000561 | NA |
| 472 | CABO-LLG-01-000860 | CTRA-DLC-01-000327 | ENZYME D-alanyl-D-alanine carboxypeptidase DacF [EC] 3.4.16.4 |
| 473 | CABO-LLG-01-000865 | CTRA-DUW-01-000710 | ENZYME Phosphoglycerate kinase [EC] 2.7.2.3 |
| 474 | CABO-LLG-01-000867 | CTRA-DUW-01-000708 | FUNCTION Phosphate transport system protein PhoU |
| 475 | CABO-LLG-01-000874 | CTRA-DUW-01-000703 | FUNCTION ABC transporter, ATP-binding protein |
| 476 | SNEG-ZXX-01-002117 | CTRA-DUW-01-000701 | FUNCTION ABC transporter, ATP-binding protein |
| 477 | CABO-LLG-01-000880 | CTRA-DUW-01-000697 | FUNCTION Ribosomal protein S2 |
| 478 | CABO-LLG-01-000881 | CTRA-DLC-01-000198 | FUNCTION Translation elongation factor EF-TS |
| 479 | CABO-LLG-01-000882 | CTRA-DUW-01-000695 | ENZYME Uridylate kinase [EC] 2.7.4. |
| 480 | CABO-LLG-01-000892 | CTRA-DUW-01-000684 | NA |
| 481 | CABO-LLG-01-000895 | CTRA-DUW-01-000681 | DOMAIN FHA |
| 482 | CABO-LLG-01-000897 | CTRA-DUW-01-000679 | ENZYME glutamyl-tRNA reductase [EC] 1.2.1. |
| 483 | CABO-LLG-01-000904 | CTRA-DUW-01-000672 | ENZYME KDO-8-phosphate synthetase [EC] 4.1.2.16 |
| 484 | CABO-LLG-01-000912 | CTRA-DLC-01-000254 | NA |
| 485 | CABO-LLG-01-000913 | CTRA-DUW-01-000640 | SIMILAR-TO Endonuclease IV [EC] 3.1.21.2 |
| 486 | CABO-LLG-01-000914 | CTRA-DUW-01-000641 | FUNCTION Ribosomal protein S4 |
| 487 | CABO-LLG-01-000916 | CTRA-DUW-01-000657 | FUNCTION Multidrug-efflux transporter |
| 488 | CABO-LLG-01-000917 | CTRA-DLC-01-000238 | ENZYME Exodeoxyribonuclease V gamma subunit [EC] 3.1.11.5 |
| 489 | CABO-LLG-01-000920 | CTRA-DUW-01-000652 | SIMILAR-TO Amino-acid aminotransferase class I [EC] 2.6.1. |
| 490 | CABO-LLG-01-000921 | CTRA-DUW-01-000651 | FUNCTION Transcription Elongation Factor GreA C-terminus |
| 491 | CABO-LLG-01-000923 | CTRA-DUW-01-000649 | NA |
| 492 | CABO-LLG-01-000924 | CTRA-DLC-01-000245 | ENZYME Porphobilinogen synthase [EC] 4.2.1.24 |
* Eight clusters do not contain one member per genome exactly and are marked; these include cluster 420 which does not contain a master sequence from the C. trachomatis original annotation dataset; NA: not available. Lead sequence is first sequence found in cluster; master sequence is sequence where annotation is drawn from (see Experimental Section for details).
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).