
Intracranial Large Artery Involvement in Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy: A Tale of Two Genes?


Abstract
1. Introduction
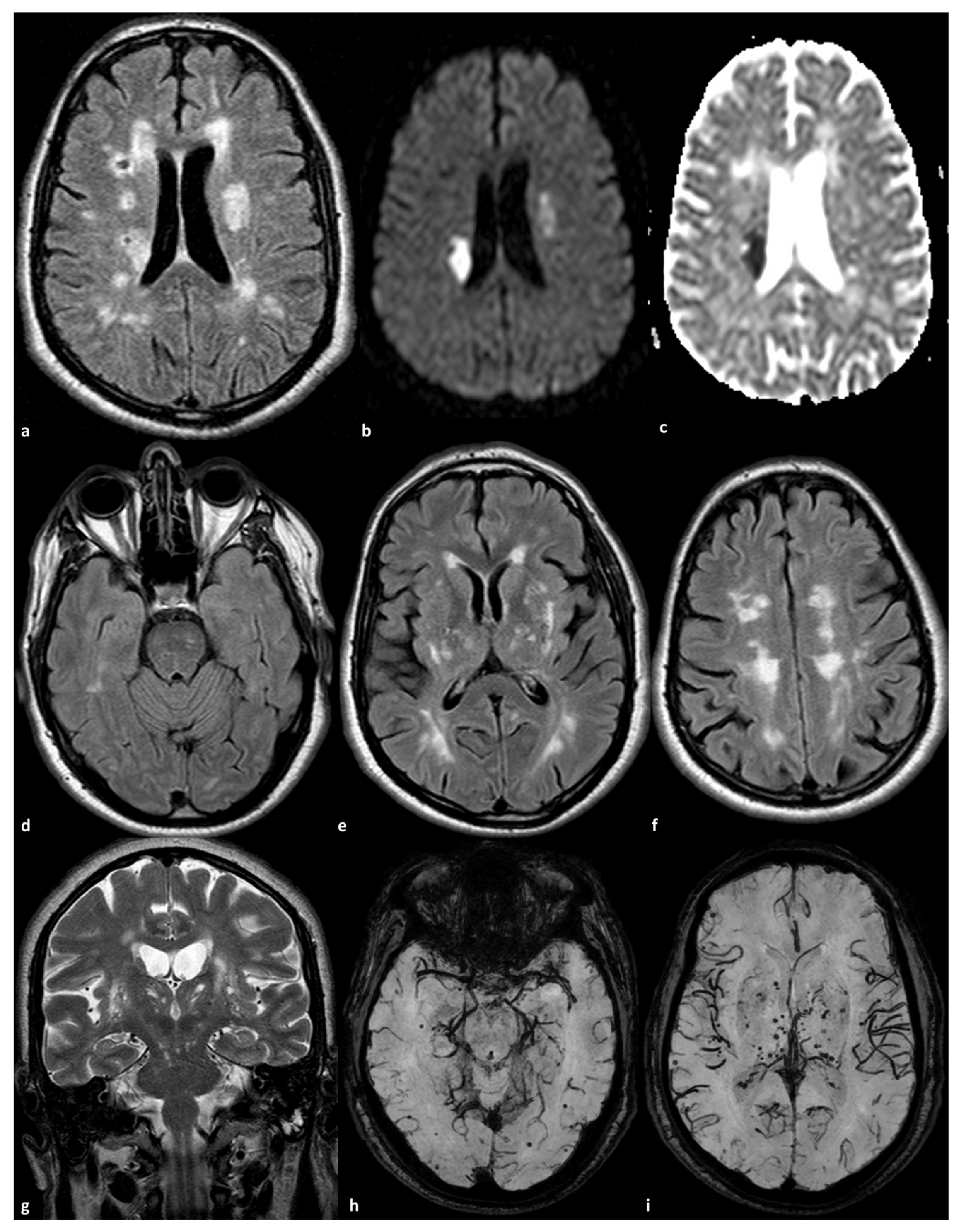
2. Intracranial Vessel Involvement in CADASIL
2.1. Small Arteries Involvement
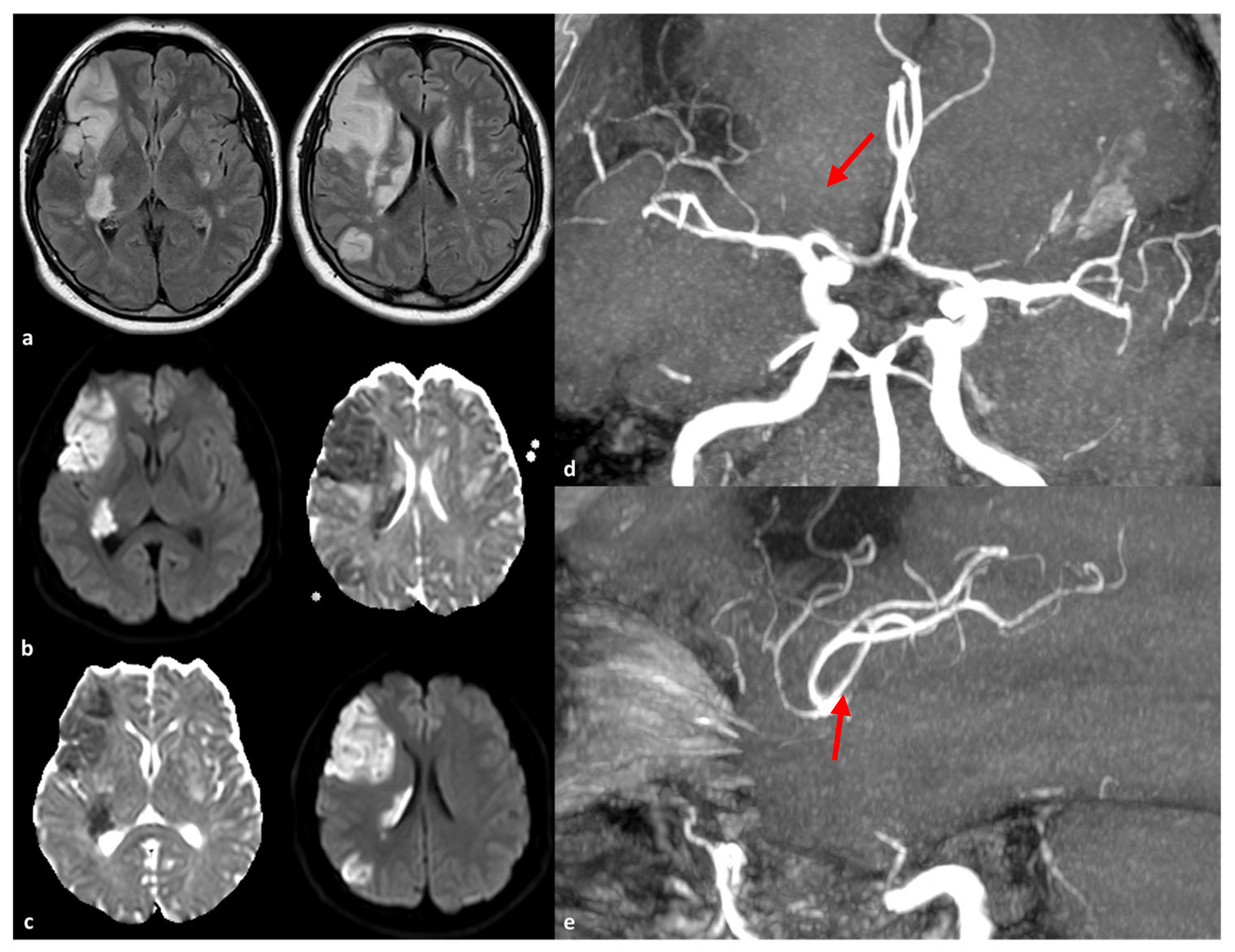
2.2. Large Artery Involvement
3. The Role of RNF213 Polymorphisms
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Choi, J.C. Genetics of cerebral small vessel disease. J. Stroke 2015, 17, 7–16. [Google Scholar] [CrossRef]
- Choi, J.C. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: A genetic cause of cerebral small vessel disease. J. Clin. Neurol. 2010, 6, 1–9. [Google Scholar] [CrossRef]
- Chabriat, H.; Joutel, A.; Dichgans, M.; Tournier-Lasserve, E.; Bousser, M.G. Cadasil. Lancet Neurol. 2009, 8, 643–653. [Google Scholar] [CrossRef]
- Tikka, S.; Mykkänen, K.; Ruchoux, M.-M.; Bergholm, R.; Junna, M.; Pöyhönen, M.; Yki-Järvinen, H.; Joutel, A.; Viitanen, M.; Baumann, M.; et al. Congruence between NOTCH3 mutations and GOM in 131 CADASIL patients. Brain 2009, 132 Pt 4, 933–939. [Google Scholar] [CrossRef]
- Joutel, A.; Corpechot, C.; Ducros, A.; Vahedi, K.; Chabriat, H.; Mouton, P.; Alamowitch, S.; Domenga, V.; Cécillion, M.; Maréchal, E.; et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 1996, 383, 707–710. [Google Scholar] [CrossRef]
- Ishiko, A.; Shimizu, A.; Nagata, E.; Takahashi, K.; Tabira, T.; Suzuki, N. Notch3 ectodomain is a major component of granular osmiophilic material (GOM) in CADASIL. Acta Neuropathol. 2006, 112, 333–339. [Google Scholar] [CrossRef]
- Joutel, A.; Haddad, I.; Ratelade, J.; Nelson, M.T. Perturbations of the cerebrovascular matrisome: A convergent mechanism in small vessel disease of the brain? J. Cereb. Blood Flow Metab. 2016, 36, 143–157. [Google Scholar] [CrossRef]
- Razvi, S.S.; Davidson, R.; Bone, I.; Muir, K.W. The prevalence of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) in the west of Scotland. J. Neurol. Neurosurg. Psychiatry 2005, 76, 739–741. [Google Scholar] [CrossRef]
- Bianchi, S.; Zicari, E.; Carluccio, A.; Di Donato, I.; Pescini, F.; Nannucci, S.; Valenti, R.; Ragno, M.; Inzitari, D.; Pantoni, L.; et al. CADASIL in central Italy: A retrospective clinical and genetic study in 229 patients. J. Neurol. 2015, 262, 134–141. [Google Scholar] [CrossRef]
- Moreton, F.C.; Razvi, S.S.; Davidson, R.; Muir, K.W. Changing clinical patterns and increasing prevalence in CADASIL. Acta Neurol. Scand. 2014, 130, 197–203. [Google Scholar] [CrossRef]
- Narayan, S.K.; Gorman, G.; Kalaria, R.N.; Ford, G.A.; Chinnery, P.F. The minimum prevalence of CADASIL in northeast England. Neurology 2012, 78, 1025–1027. [Google Scholar] [CrossRef]
- Rutten, J.W.; Dauwerse, H.G.; Gravesteijn, G.; van Belzen, M.J.; van der Grond, J.; Polke, J.M.; Bernal-Quiros, M.; Oberstein, S.A.J.L. Archetypal NOTCH3 mutations frequent in public exome: Implications for CADASIL. Ann. Clin. Transl. Neurol. 2016, 3, 844–853. [Google Scholar] [CrossRef]
- Grami, N.; Chong, M.; Lali, R.; Mohammadi-Shemirani, P.; Henshall, D.E.; Rannikmäe, K.; Paré, G. Global assessment of Mendelian stroke genetic prevalence in 101 635 individuals from 7 ethnic groups. Stroke 2020, 51, 1290–1293. [Google Scholar] [CrossRef]
- Choi, J.C.; Kang, S.Y.; Kang, J.H.; Park, J.K. Intracerebral hemorrhages in CADASIL. Neurology 2006, 67, 2042–2044. [Google Scholar] [CrossRef]
- Liao, Y.-C.; Hsiao, C.-T.; Fuh, J.-L.; Chern, C.-M.; Lee, W.-J.; Guo, Y.-C.; Wang, S.-J.; Lee, I.-H.; Liu, Y.-T.; Wang, Y.-F.; et al. Characterization of CADASIL among the Han Chinese in Taiwan: Distinct genotypic and phenotypic profiles. PLoS ONE 2015, 10, e0136501. [Google Scholar] [CrossRef]
- Dichgans, M.; Mayer, M.; Uttner, I.; Brüning, R.; Müller-Höcker, J.; Rungger, G.; Ebke, M.; Klockgether, T.; Gasser, T. The phenotypic spectrum of CADASIL: Clinical findings in 102 cases. Ann. Neurol. 1998, 44, 731–739. [Google Scholar] [CrossRef]
- Liem, M.K.; van der Grond, J.; Haan, J.; Boom, R.v.D.; Ferrari, M.D.; Knaap, Y.M.; Breuning, M.H.; van Buchem, M.A.; Middelkoop, H.A.; Oberstein, S.A.L. Lacunar infarcts are the main correlate with cognitive dysfunction in CADASIL. Stroke 2007, 38, 923–928. [Google Scholar] [CrossRef]
- O’Sullivan, M.; Jarosz, J.M.; Martin, R.J.; Deasy, N.; Powell, J.F.; Markus, H.S. MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology 2001, 56, 628–634. [Google Scholar] [CrossRef]
- Chabriat, H.; Hervé, D.; Duering, M.; Godin, O.; Jouvent, E.; Opherk, C.; Alili, N.; Reyes, S.; Jabouley, A.; Zieren, N.; et al. Predictors of clinical worsening in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: Prospective cohort study. Stroke 2016, 47, 4–11. [Google Scholar] [CrossRef]
- Jickling, G.C.; Sharp, F.R. Biomarker panels in ischemic stroke. Stroke 2015, 46, 915–920. [Google Scholar] [CrossRef]
- Vilar-Bergua, A.; Riba-Llena, I.; Nafría, C.; Bustamante, A.; Llombart, V.; Delgado, P.; Montaner, J. Blood and CSF biomarkers in brain subcortical ischemic vascular disease: Involved pathways and clinical applicability. J. Cereb. Blood Flow Metab. 2016, 36, 55–71. [Google Scholar] [CrossRef]
- Baudrimont, M.; Dubas, F.; Joutel, A.; Tournier-Lasserve, E.; Bousser, M.G. Autosomal dominant leukoencephalopathy and subcortical ischemic stroke. A clinicopathological study. Stroke 1993, 24, 122–125. [Google Scholar] [CrossRef]
- Ruchoux, M.M.; Guerouaou, D.; Vandenhaute, B.; Pruvo, J.P.; Vermersch, P.; Leys, D. Systemic vascular smooth muscle cell impairment in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Acta Neuropathol. 1995, 89, 500–512. [Google Scholar] [CrossRef]
- Ruchoux, M.M.; Domenga, V.; Brulin, P.; Maciazek, J.; Limol, S.; Tournier-Lasserve, E.; Joutel, A. Transgenic mice expressing mutant Notch3 develop vascular alterations characteristic of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Am. J. Pathol. 2003, 162, 329–342. [Google Scholar] [CrossRef]
- Choi, J.C.; Song, S.K.; Lee, J.S.; Kang, S.Y.; Kang, J.H. Diversity of stroke presentation in CADASIL: Study from patients harboring the predominant NOTCH3 mutation R544C. J. Stroke Cerebrovasc. Dis. 2013, 22, 126–131. [Google Scholar] [CrossRef]
- Kang, H.G.; Kim, J.S. Intracranial arterial disease in CADASIL patients. J. Neurol. Sci. 2015, 359, 347–350. [Google Scholar] [CrossRef]
- Gutierrez, J.; Morgello, S.; Dwork, A.; Brickman, A.; Mohr, J.P.; Elkind, M.S.; Marshall, R. Relationship between parent brain large arteries and their penetrating arterial offspring. Stroke 2019, 50, ATMP103. [Google Scholar] [CrossRef]
- Romay, M.C.; Knutsen, R.H.; Ma, F.; Mompeón, A.; Hernandez, G.E.; Salvador, J.; Mirkov, S.; Batra, A.; Sullivan, D.P.; Procissi, D.; et al. Age-related loss of Notch3 underlies brain vascular contractility deficiencies, glymphatic dysfunction, and neurodegeneration in mice. J. Clin. Investig. 2024, 134, e166134. [Google Scholar] [CrossRef]
- Yeung, W.T.E.; Mizuta, I.; Watanabe-Hosomi, A.; Yokote, A.; Koizumi, T.; Mukai, M.; Kinoshita, M.; Ohara, T.; Mizuno, T. RNF213-related susceptibility of Japanese CADASIL patients to intracranial arterial stenosis. J. Hum. Genet. 2018, 63, 687–690. [Google Scholar] [CrossRef]
- Zedde, M.; Grisendi, I.; Assenza, F.; Napoli, M.; Moratti, C.; Pavone, C.; Bonacini, L.; Di Cecco, G.; D’aniello, S.; Stoenoiu, M.S.; et al. RNF213 Polymorphisms in Intracranial Artery Dissection. Genes 2024, 15, 725. [Google Scholar] [CrossRef]
- Kim, H.J.; Choi, E.H.; Chung, J.W.; Kim, J.H.; Kim, Y.S.; Seo, W.K.; Kim, G.M.; Bang, O.Y. Role of the RNF213 Variant in Vascular Outcomes in Patients With Intracranial Atherosclerosis. J. Am. Heart Assoc. 2021, 10, e017660. [Google Scholar] [CrossRef]
- Kalimo, H.; Ruchoux, M.M.; Viitanen, M.; Kalaria, R.N. CADASIL: A common form of hereditary arteriopathy causing brain infarcts and dementia. Brain Pathol. 2002, 12, 371–384. [Google Scholar] [CrossRef]
- Chabriat, H.; Vahedi, K.; Iba-Zizen, M.T.; Joutel, A.; Nibbio, A.; Nagy, T.G.; Krebs, M.O.; Julien, J.; Dubois, B.; Ducrocq, X.; et al. Clinical spectrum of CADASIL: A study of 7 families. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Lancet 1995, 346, 934–939. [Google Scholar] [CrossRef]
- Chabriat, H.; Levy, C.; Taillia, H.; Iba-Zizen, M.T.; Vahedi, K.; Joutel, A.; Tournier-Lasserve, E.; Bousser, M.G. Patterns of MRI lesions in CADASIL. Neurology 1998, 51, 452–457. [Google Scholar] [CrossRef]
- Chabriat, H.; Pappata, S.; Poupon, C.; Clark, C.A.; Vahedi, K.; Poupon, F.; Mangin, J.F.; Pachot-Clouard, M.; Jobert, A.; Le Bihan, D.; et al. Clinical severity in CADASIL related to ultrastructural damage in white matter: In vivo study with diffusion tensor MRI. Stroke 1999, 30, 2637–2643. [Google Scholar] [CrossRef]
- Skehan, S.J.; Hutchinson, M.; MacErlaine, D.P. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: MR findings. Am. J. Neuroradiol. 1995, 16, 2115–2119. [Google Scholar]
- Gutierrez-Molina, M.; Caminero Rodriguez, A.; Martinez Garcia, C.; Arpa Gutierrez, J.; Morales Bastos, C.; Amer, G. Small arterial granular degeneration in familial Binswanger’s syndrome. Acta Neuropathol. 1994, 87, 98–105. [Google Scholar] [CrossRef]
- Sourander, P.; Walinder, J. Hereditary multi-infarct dementia. Morphological and clinical studies of a new disease. Acta Neuropathol. 1977, 39, 247–254. [Google Scholar] [CrossRef]
- Brulin, P.; Godfraind, C.; Leteurtre, E.; Ruchoux, M.M. Morphometric analysis of ultrastructural vascular changes in CADASIL: Analysis of 50 skin biopsy specimens and pathogenic implications. Acta Neuropathol. 2002, 104, 241–248. [Google Scholar] [CrossRef]
- Miao, Q.; Paloneva, T.; Tuominen, S.; Pöyhönen, M.; Tuisku, S.; Viitanen, M.; Kalimo, H. Fibrosis and stenosis of the long penetrating cerebral arteries: The cause of the white matter pathology in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Brain Pathol. 2004, 14, 358–364. [Google Scholar] [CrossRef]
- Furuta, A.; Ishii, N.; Nishihara, Y.; Horie, A. Medullary arteries in aging and dementia. Stroke 1991, 22, 442–446. [Google Scholar] [CrossRef]
- Lammie, G.A.; Brannan, F.; Slattery, J.; Warlow, C. Nonhypertensive cerebral small-vessel disease. An autopsy study. Stroke 1997, 28, 2222–2229. [Google Scholar] [CrossRef]
- Okeda, R.; Arima, K.; Kawai, M. Arterial changes in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) in relation to pathogenesis of diffuse myelin loss of cerebral white matter: Examination of cerebral medullary arteries by reconstruction of serial sections of an autopsy case. Stroke 2002, 33, 2565–2569. [Google Scholar]
- Vinters, H.V.; Gilbert, J.J. Cerebral amyloid angiopathy: Incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 1983, 14, 924–928. [Google Scholar] [CrossRef]
- Bruening, R.; Dichgans, M.; Berchtenbreiter, C.; Yousry, T.; Seelos, K.C.; Wu, R.H.; Mayer, M.; Brix, G.; Reiser, M. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: Decrease in regional cerebral blood volume in hyperintense subcortical lesions inversely correlates with disability and cognitive performance. Am. J. Neuroradiol. 2001, 22, 1268–1274. [Google Scholar]
- Chabriat, H.; Pappata, S.; Ostergaard, L.; Clark, C.A.; Pachot-Clouard, M.; Vahedi, K.; Jobert, A.; Le Bihan, D.; Bousser, M.G. Cerebral hemodynamics in CADASIL before and after acetazolamide challenge assessed with MRI bolus tracking. Stroke 2000, 31, 1904–1912. [Google Scholar] [CrossRef]
- Tuominen, S.; Miao, Q.; Kurki, T.; Tuisku, S.; Pöyhönen, M.; Kalimo, H.; Viitanen, M.; Rinne, J.O. Cerebral blood flow and glucose metabolism studies on young CADASIL subjects. Stroke 2004, 35, 1063–1067. [Google Scholar] [CrossRef]
- Pfefferkorn, T.; von Stuckrad-Barre, S.; Herzog, J.; Gasser, T.; Hamann, G.F.; Dichgans, M. Reduced cerebrovascular CO2 reactivity in CADASIL: A transcranial Doppler sonography study. Stroke 2001, 32, 17–21. [Google Scholar] [CrossRef]
- Liebetrau, M.; Herzog, J.; Kloss, C.U.; Hamann, G.F.; Dichgans, M. Prolonged cerebral transit time in CADASIL: A transcranial ultrasound study. Stroke 2002, 33, 509–512. [Google Scholar] [CrossRef]
- Van Den Boom, R.; LesnikOberstein, S.A.; Spilt, A.; Behloul, F.; Ferrari, M.D.; Haan, J.; Westendorp, R.G.; Van Buchem, M.A. Cerebral hemodynamics and white matter hyperintensities in CADASIL. J. Cereb. Blood Flow Metab. 2003, 23, 599–604. [Google Scholar] [CrossRef]
- Ling, C.; Fang, X.; Kong, Q.; Sun, Y.; Wang, B.; Zhuo, Y.; An, J.; Zhang, W.; Wang, Z.; Zhang, Z.; et al. Lenticulostriate Arteries and Basal Ganglia Changes in Cerebral Autosomal Dominant Arteriopathy With Subcortical Infarcts and Leukoencephalopathy, a High-Field MRI Study. Front. Neurol. 2019, 10, 870. [Google Scholar] [CrossRef]
- Miao, Q.; Paloneva, T.; Tuisku, S.; Roine, S.; Poyhonen, M.; Viitanen, M.; Kalimo, H. Arterioles of the lenticular nucleus in CADASIL. Stroke 2006, 37, 2242–2247. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Ihara, M.; Tham, C.; Low, R.W.; Slade, J.Y.; Moss, T.; Oakley, A.E.; Polvikoski, T.; Kalaria, R.N. Neuropathological correlates of temporal pole white matter hyperintensities in CADASIL. Stroke 2009, 40, 2004–2011. [Google Scholar] [CrossRef]
- Lopez-Navarro, E.R.; Mayer, S.V.; Barreto, B.R.; Strobino, K.H.; Spagnolo-Allende, A.; Bueno, P.G.; Gurel, K.; Kozii, K.; Rahman, S.; Khasiyev, F.; et al. Assessing changes on large cerebral arteries in CADASIL: Preliminary insights from a case-control analysis. J. Stroke Cerebrovasc. Dis. 2025, 34, 108294. [Google Scholar] [CrossRef]
- Santa, Y.; Uyama, E.; Chui, D.H.; Arima, M.; Kotorii, S.; Takahashi, K.; Tabira, T. Genetic, clinical and pathological studies of cadasil in Japan: A partial contribution of notch3 mutations and implications of smooth muscle cell degeneration for the pathogenesis. J. Neurol. Sci. 2003, 212, 79–84. [Google Scholar] [CrossRef]
- Nishio, T.; Arima, K.; Eto, K.; Ogawa, M.; Sunohara, N. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy—Report of an autopsied Japanese case. Clin. Neurol. 1997, 37, 910–916. [Google Scholar]
- Choi, E.J.; Choi, C.G.; Kim, J.S. Large cerebral artery involvement in cadasil. Neurology 2005, 65, 1322–1324. [Google Scholar] [CrossRef]
- Engelter, S.T.; Rueegg, S.; Kirsch, E.C.; Fluri, F.; Probst, A.; Steck, A.J.; Lyrer, P.A. CADASIL mimicking primary angiitis of the central nervous system. Arch. Neurol. 2002, 59, 1480–1483. [Google Scholar] [CrossRef]
- Li, H.; Wong, K.S. Racial distribution of intracranial and extracranial atherosclerosis. J. Clin. Neurosci. 2003, 10, 30–34. [Google Scholar] [CrossRef]
- Wong, K.S.; Huang, Y.N.; Gao, S.; Lam, W.; Chan, Y.L.; Kay, R. Intracranial stenosis in Chinese patients with acute stroke. Neurology 1998, 50, 812–813. [Google Scholar] [CrossRef]
- Bae, H.-J.; Lee, J.; Park, J.-M.; Kwon, O.; Koo, J.-S.; Kim, B.-K.; Pandey, D.K. Risk factors of intracranial cerebral atherosclerosis among asymptomatics. Cerebrovasc. Dis. 2007, 24, 355–360. [Google Scholar] [CrossRef]
- Jo, I.; Ahn, Y.; Lee, J.; Shin, K.R.; Lee, H.K.; Shin, C. Prevalence, awareness, treatment, control and risk factors of hypertension in Korea: The ANASAN Study. J. Hypertens. 2001, 19, 1523–1532. [Google Scholar] [CrossRef]
- Singhal, S.; Bevan, S.; Barrick, T.; Rich, P.; Markus, H.S. The influence of genetic and cardiovascular risk factors on the CADASIL phenotype. Brain 2004, 127, 2031–2038. [Google Scholar] [CrossRef]
- Yin, X.; Wu, D.; Wan, J.; Yan, S.; Lou, M.; Zhao, G.; Zhang, B. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: Phenotypic and mutational spectrum in patients from mainland China. Int. J. Neurosci. 2014, 125, 585–592. [Google Scholar] [CrossRef]
- Lee, Y.C.; Liu, C.S.; Chang, M.H.; Lin, K.P.; Fuh, J.L.; Lu, Y.C.; Liu, Y.F.; Soong, B.W. Population-specific spectrum of NOTCH3 mutations, MRI features and founder effect of CADASIL in Chinese. J. Neurol. 2009, 256, 249–255. [Google Scholar] [CrossRef]
- Zhang, C.; Li, W.; Li, S.; Niu, S.; Wang, X.; Yu, X.; Zhang, Z. Intracranial Large Artery Abnormalities and Association with Cerebral Small Vessel Disease in CADASIL. Front. Neurol. 2020, 11, 726. [Google Scholar] [CrossRef]
- Kim, J.S.; Nah, H.W.; Park, S.M.; Kim, S.K.; Cho, K.H.; Lee, J.; Lee, Y.S.; Kim, J.; Ha, S.W.; Kim, E.G. Risk factors and stroke mechanisms in atherosclerotic stroke: Intracranial compared with extracranial and anterior compared with posterior circulation disease. Stroke 2012, 43, 3313–3318. [Google Scholar] [CrossRef]
- Tan, S.M.L.; Chin, H.L.; Makmur, A.; Tan, B.Y.Q. Medium and Large Artery Aneurysms in Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy. Ann. Neurol. 2024, 96, 726–728. [Google Scholar] [CrossRef]
- Agarwal, A.; Mazumdar, P.; Gupta, P.; Garg, A.; Radhakrishnan, D.M.; Das, A.; Pandit, A.K.; Srivastava, A.K. Fusiform Intracranial Aneurysms in a CADASIL Patient: A Possibly Missed Association. Ann. Indian Acad. Neurol. 2022, 25, 747–749. [Google Scholar] [CrossRef]
- Haas, T.L.; Doyle, J.L.; Distasi, M.R.; Norton, L.E.; Sheridan, K.M.; Unthank, J.L. Involvement of MMPs in the outward remodeling of collateral mesenteric arteries. Am. J. Physiol. Heart Circulat. Physiol. 2007, 293, H2429–H2437. [Google Scholar] [CrossRef]
- Gasser, T.C.; Ogden, R.W.; Holzapfel, G.A. Hyperelastic modelling of arterial layers with distributed collagen fibre orientations. J. R. Soc. Interface 2006, 3, 15–35. [Google Scholar] [CrossRef]
- Gutierrez, J.; Elkind, M.S.V.; Petito, C.; Chung, D.Y.; Dwork, A.J.; Marshall, R.S. The contribution of HIV infection to intracranial arterial remodeling: A pilot study. Neuropathology 2013, 33, 256–263. [Google Scholar] [CrossRef]
- Sho, E.; Nanjo, H.; Sho, M.; Kobayashi, M.; Komatsu, M.; Kawamura, K.; Xu, C.; Zarins, C.K.; Masuda, H. Arterial enlargement, tortuosity, and intimal thickening in response to sequential exposure to high and low wall shear stress. J. Vasc. Surg. 2004, 39, 601–612. [Google Scholar] [CrossRef]
- Gutierrez, J.; Menshawy, K.; Goldman, J.; Dwork, A.J.; Elkind, M.S.; Marshall, R.S.; Morgello, S. Metalloproteinases and brain arterial remodeling among individuals with and those without HIV infection. J. Infect. Dis. 2016, 214, 1329–1335. [Google Scholar] [CrossRef]
- Gutierrez, J.; Menshawy, K.; Gonzalez, M.; Goldman, J.; Elkind, M.S.; Marshall, R.; Morgello, S. Brain large artery inflammation associated with HIV and large artery remodeling. AIDS 2016, 30, 415–423. [Google Scholar] [CrossRef]
- Lacoste, B.; Comin, C.H.; Ben-Zvi, A.; Kaeser, P.S.; Xu, X.; Costa Lda, F.; Gu, C. Sensory-related neural activity regulates the structure of vascular networks in the cerebral cortex. Neuron 2014, 83, 1117–1130. [Google Scholar] [CrossRef]
- Roth, W.; Morgello, S.; Goldman, J.; Mohr, J.P.; Elkind, M.S.; Marshall, R.S.; Gutierrez, J. Histopathological differences between the anterior and posterior brain arteries as a function of aging. Stroke 2017, 48, 638–644. [Google Scholar] [CrossRef]
- Smith, A.S.; Bellon, J.R. Parallel and spiral flow patterns of vertebral artery contributions to the basilar artery. AJNR Am. J. Neuroradiol. 1995, 16, 1587–1591. [Google Scholar]
- Gutierrez, J.; Bagci, A.; Del Brutto, V.; Gervasi-Franklin, P.; Rundek, T.; Elkind, M.S.; Alperin, N.; Sacco, R.L.; Wright, C.B. Correlates of dolichoectasia in an urban, stroke-free cohort: Results from the Northern Manhattan study. Stroke 2014, 45, ATP152. [Google Scholar] [CrossRef]
- Del Brutto, V.J.; Khasiyev, F.; Liu, M.; Spagnolo-Allende, A.; Qiao, Y.; Melgarejo Arias, J.D.; Guzman, V.A.; Igwe, K.C.; Sanchez, D.L.; Andrews, H.; et al. Association of brain arterial diameters with demographic and anatomical factors in a multi-national pooled analysis of cohort studies. Neuroradiol. J. 2024, 37, 304–313. [Google Scholar] [CrossRef]
- Koizumi, A.; Kobayashi, H.; Hitomi, T.; Harada, K.H.; Habu, T.; Youssefian, S. A new horizon of moyamoya disease and associated health risks explored through RNF213. Environ. Health Prev. Med. 2016, 21, 55–70. [Google Scholar] [CrossRef]
- Liu, W.; Morito, D.; Takashima, S.; Mineharu, Y.; Kobayashi, H.; Hitomi, T.; Hashikata, H.; Matsuura, N.; Yamazaki, S.; Toyoda, A.; et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS ONE 2011, 6, e22542. [Google Scholar] [CrossRef]
- Hashimoto, N.; Tominaga, T.; Miyamoto, S.; Nagata, I.; Houkin, K.; Suzuki, N.; Takagi, Y. Research Committee on the Pathology and Treatment of Spontaneous Occlusion of the Circle of Willis; Health Labour Sciences Research Grant for Research on Measures for Infractable Diseases. Guidelines for diagnosis and treatment of moyamoya disease (spontaneous occlusion of the circle of Willis). Neurol. Med. Chir. 2012, 52, 245–266. [Google Scholar]
- Kamada, F.; Aoki, Y.; Narisawa, A.; Abe, Y.; Komatsuzaki, S.; Kikuchi, A.; Kanno, J.; Niihori, T.; Ono, M.; Ishii, N.; et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J. Hum. Genet. 2011, 56, 34–40. [Google Scholar] [CrossRef]
- Lin, J.; Sheng, W. RNF213 variant diversity predisposes distinct populations to dissimilar cerebrovascular diseases. BioMed Res. Int. 2018, 2018, 6359174. [Google Scholar] [CrossRef]
- Sun, X.S.; Wen, J.; Li, J.X.; Lai, R.; Wang, Y.-F.; Liu, H.-J.; Sheng, W.-L. The association between the ring finger protein 213 (RNF213) polymorphisms and moyamoya disease susceptibility: A meta-analysis based on case-control studies. Mol. Genet. Genom. 2016, 291, 1193–1203. [Google Scholar] [CrossRef]
- Guey, S.; Kraemer, M.; Hervé, D.; Ludwig, T.; Kossorotoff, M.; Bergametti, F.; Schwitalla, J.C.; Choi, S.; Broseus, L.; Callebaut, I.; et al. Rare RNF213 variants in the C-terminal region encompassing the RING-finger domain are associated with moyamoya angiopathy in Caucasians. Eur. J. Hum. Genet. 2017, 25, 995–1003. [Google Scholar] [CrossRef]
- Miyawaki, S.; Imai, H.; Shimizu, M.; Yagi, S.; Ono, H.; Mukasa, A.; Nakatomi, H.; Shimizu, T.; Saito, N. Genetic variant RNF213 c.14576G>A in various phenotypes of intracranial major artery stenosis/occlusion. Stroke 2013, 44, 2894–2897. [Google Scholar] [CrossRef]
- Liao, X.; Deng, J.; Dai, W.; Zhang, T.; Yan, J. Rare variants of RNF213 and moyamoya/non-moyamoya intracranial artery stenosis/occlusion disease risk: A meta-analysis and systematic review. Environ. Health Prev. Med. 2017, 22, 75. [Google Scholar] [CrossRef]
- Ishisaka, E.; Watanabe, A.; Murai, Y.; Shirokane, K.; Matano, F.; Tsukiyama, A.; Baba, E.; Nakagawa, S.; Tamaki, T.; Mizunari, T.; et al. Role of RNF213 polymorphism in defining quasi-moyamoya disease and definitive moyamoya disease. Neurosurg. Focus 2021, 51, E2. [Google Scholar] [CrossRef]
- Fukushima, H.; Takenouchi, T.; Kosaki, K. Homozygosity for moyamoya disease risk allele leads to moyamoya disease with extracranial systemic and pulmonary vasculopathy. Am. J. Med. Genet. A 2016, 170, 2453–2456. [Google Scholar] [CrossRef]
- Bang, O.Y.; Chung, J.W.; Kim, D.H.; Won, H.H.; Yeon, J.Y.; Ki, C.S.; Shin, H.J.; Kim, J.S.; Hong, S.C.; Kim, D.K.; et al. Moyamoya Disease and Spectrums of RNF213 Vasculopathy. Transl. Stroke Res. 2020, 11, 580–589. [Google Scholar] [CrossRef]
- Suzuki, H.; Kataoka, M.; Hiraide, T.; Aimi, Y.; Yamada, Y.; Katsumata, Y.; Chiba, T.; Kanekura, K.; Isobe, S.; Sato, Y.; et al. Genomic comparison with supercentenarians identifies RNF213 as a risk gene for pulmonary arterial hypertension. Circ. Genom. Precis. Med. 2018, 11, e002317. [Google Scholar]
- Hiraide, T.; Suzuki, H.; Momoi, M.; Shinya, Y.; Fukuda, K.; Kosaki, K.; Kataoka, M. RNF213-Associated Vascular Disease: A Concept Unifying Various Vasculopathies. Life 2022, 12, 555. [Google Scholar] [CrossRef]
- Chang, S.A.; Song, J.S.; Park, T.K.; Yang, J.H.; Kwon, W.C.; Kim, S.R.; Kim, S.R.; Kim, S.M.; Cha, J.; Jang, S.Y.; et al. Nonsyndromic peripheral pulmonary artery stenosis is associated with homozygosity of RNF213 p.Arg4810Lys regardless of cooccurrence of moyamoya disease. Chest 2018, 153, 404–413. [Google Scholar] [CrossRef]
- Shinya, Y.; Miyawaki, S.; Imai, H.; Hongo, H.; Ono, H.; Takenobu, A.; Nakatomi, H.; Teraoka, A.; Saito, N. Genetic Analysis of Ring Finger Protein 213 (RNF213) c.14576G>A in Intracranial Atherosclerosis of the Anterior and Posterior Circulations. J. Stroke Cerebrovasc. Dis. 2017, 26, 2638–2644. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Nie, F.; Li, Q.; Zhang, K.; Liu, M.; Yang, L.; Zhang, Q.; Liu, S.; Zeng, F.; et al. Association of Genetic Variants with Moyamoya Disease in 13,000 Individuals: A Meta-Analysis. Stroke 2020, 51, 1647–1655. [Google Scholar] [CrossRef]
- Zhou, S.; Ambalavanan, A.; Rochefort, D.; Xie, P.; Bourassa, C.V.; Hince, P.; Dionne-Laporte, A.; Spiegelman, D.; Gan-Or, Z.; Mirarchi, C.; et al. RNF213 Is Associated with Intracranial Aneurysms in the French-Canadian Population. Am. J. Hum. Genet. 2016, 99, 1072–1085. [Google Scholar] [CrossRef]
- Miyawaki, S.; Imai, H.; Takayanagi, S.; Mukasa, A.; Nakatomi, H.; Saito, N. Identification of a genetic variant common to moyamoya disease and intracranial major artery stenosis/occlusion. Stroke 2012, 43, 3371–3374. [Google Scholar] [CrossRef]
- UCAS Japan Investigators. The Natural Course of Unruptured Cerebral Aneurysms in a Japanese Cohort. N. Engl. J. Med. 2012, 366, 2474–2482. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, J. Neoplastic cerebral aneurysm from metastatic tumor: A systematic review of clinical and treatment characteristics. Clin. Neurol. Neurosurg. 2015, 128, 107–111. [Google Scholar] [CrossRef]
- Uehara, T.; Tabuchi, M.; Mori, E. Frequency and clinical correlates of occlusive lesions of cerebral arteries in Japanese patients without stroke. Cerebrovasc. Dis. 1998, 8, 267–272. [Google Scholar] [CrossRef]
- Saito, S.; Hosoki, S.; Yamaguchi, E.; Ishiyama, H.; Abe, S.; Yoshimoto, T.; Tanaka, T.; Hattori, Y.; Liao, Y.C.; Lee, Y.C.; et al. Blended Phenotype of NOTCH3 and RNF213 Variants With Accelerated Large and Small Artery Crosstalk: A Case Report and Literature Review. Neurol. Genet. 2024, 10, e200176. [Google Scholar] [CrossRef]
- Ihara, M.; Yamamoto, Y.; Hattori, Y.; Liu, W.; Kobayashi, H.; Ishiyama, H.; Yoshimoto, T.; Miyawaki, S.; Clausen, T.; Bang, O.Y.; et al. Moyamoya disease: Diagnosis and interventions. Lancet Neurol. 2022, 21, 747–758. [Google Scholar] [CrossRef]
- Okazaki, S.; Morimoto, T.; Kamatani, Y.; Kamimura, T.; Kobayashi, H.; Harada, K.; Tomita, T.; Higashiyama, A.; Takahashi, J.C.; Nakagawara, J.; et al. Moyamoya disease susceptibility variant RNF213 p.R4810K increases the risk of ischemic stroke attributable to large-artery atherosclerosis. Circulation 2019, 139, 295–298. [Google Scholar] [CrossRef]
- Zedde, M.; Pascarella, R. ABCC6 Involvement in Cerebral Small Vessel Disease: Potential Mechanisms and Associations. Genes 2025, 16, 728. [Google Scholar] [CrossRef]
- Bersano, A.; Khan, N.; Fuentes, B.; Acerbi, F.; Canavero, I.; Tournier-Lasserve, E.; Vajcoczy, P.; Zedde, M.L.; Hussain, S.; Lémeret, S.; et al. European Stroke Organisation (ESO) Guidelines on Moyamoya angiopathy Endorsed by Vascular European Reference Network (VASCERN). Eur. Stroke J. 2023, 8, 55–84. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| RNF213 Variant | Reference | Cases (n) | Controls (n) | Country | Main Findings |
|---|---|---|---|---|---|
| c.14576G > A | 29 | 124 | 384 | Japan | Carrier rate in CADASIL patients with intracranial stenosis: 23.5% (4/17) vs. 1.9% (2/107) without stenosis (p = 0.0032). Higher frequency of territorial infarction in carriers (75.0% vs. 20.0%, p = 0.0410). 2.6% of control population identified as carriers of the c.14576G > A variant. 21.9% of sporadic intracranial stenosis patients exhibited this variant in the Japanese population [5]. |
| c.14429G > A (p.Arg4810Lys) | 29 | 124 | 384 | Japan | Found in 4.8% (6/124) of CADASIL patients; higher than 1.5% in the general population. Present in 80–90% of familial moyamoya disease cases; significant risk factor for intracranial artery stenosis [12]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zedde, M.; Pascarella, R. Intracranial Large Artery Involvement in Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy: A Tale of Two Genes? Genes 2025, 16, 882. https://doi.org/10.3390/genes16080882
Zedde M, Pascarella R. Intracranial Large Artery Involvement in Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy: A Tale of Two Genes? Genes. 2025; 16(8):882. https://doi.org/10.3390/genes16080882
Chicago/Turabian StyleZedde, Marialuisa, and Rosario Pascarella. 2025. "Intracranial Large Artery Involvement in Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy: A Tale of Two Genes?" Genes 16, no. 8: 882. https://doi.org/10.3390/genes16080882
APA StyleZedde, M., & Pascarella, R. (2025). Intracranial Large Artery Involvement in Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy: A Tale of Two Genes? Genes, 16(8), 882. https://doi.org/10.3390/genes16080882