
Genetic Landscape of Familial Melanoma



Abstract
1. Introduction
2. Methods
3. Defining Familial Melanoma Concept
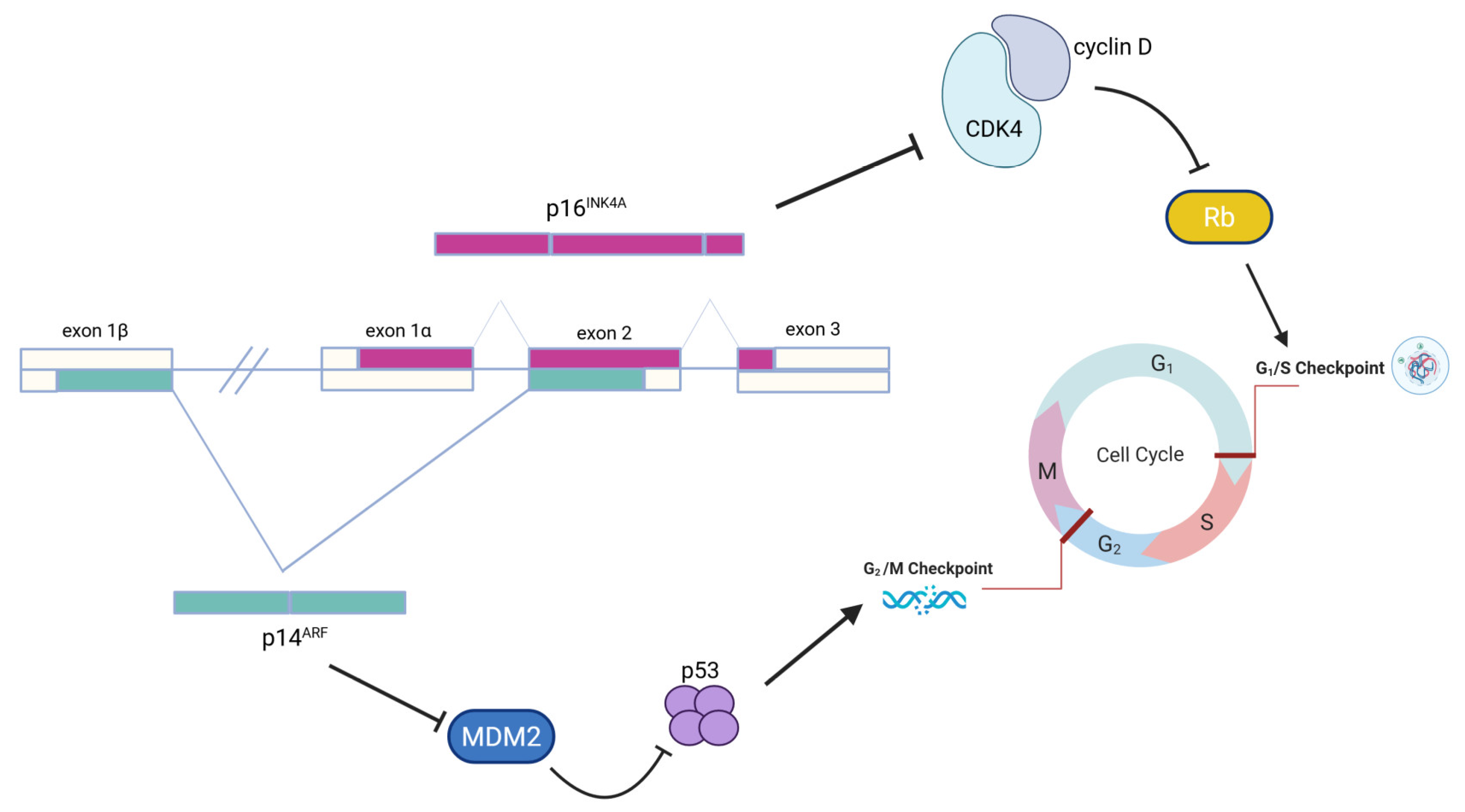
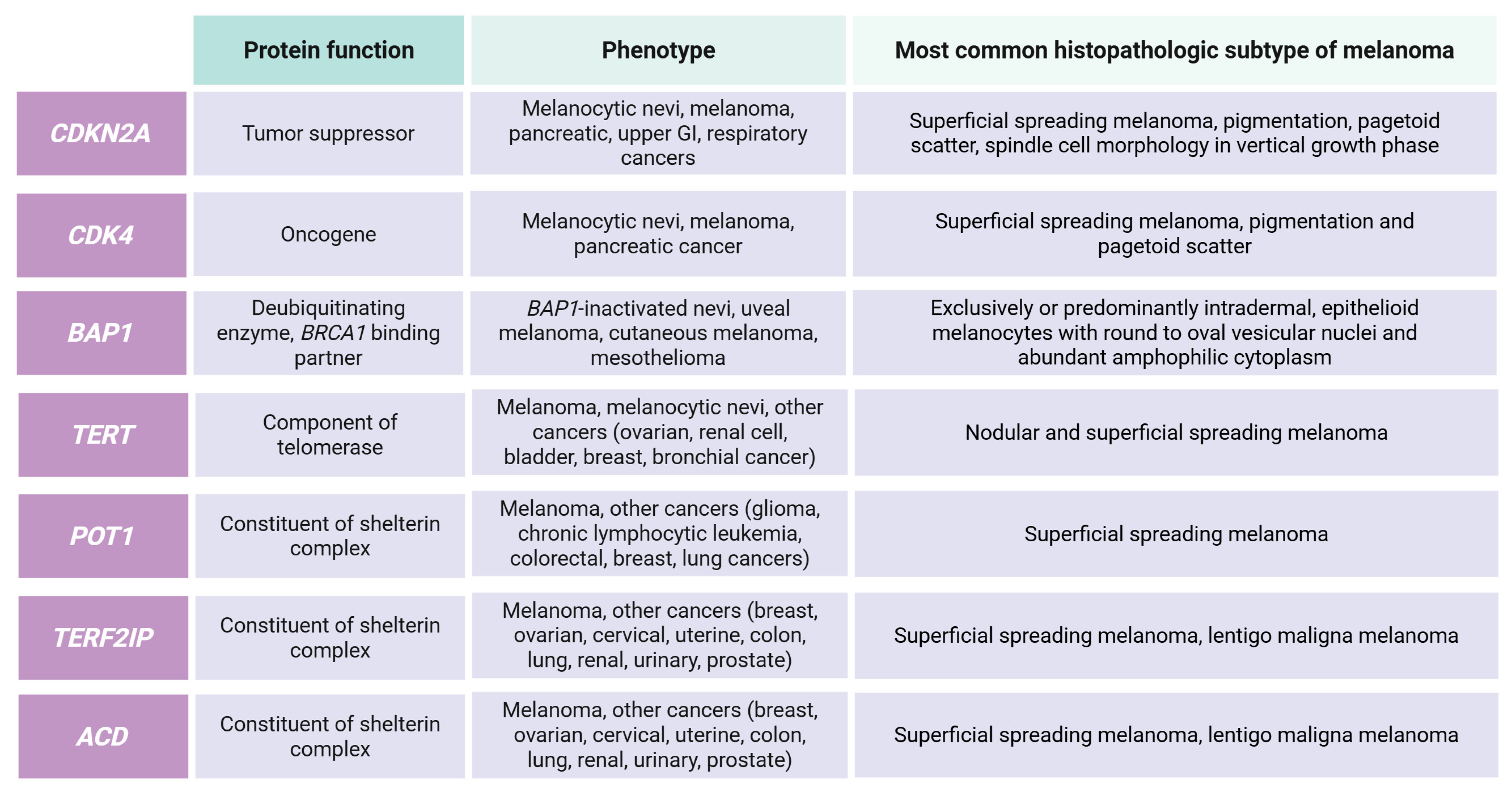
3.1. High-Risk Genes
3.2. Moderate-Risk Genes
3.3. Low-Risk Genes
3.4. Candidate Genes
3.5. Susceptibility Loci
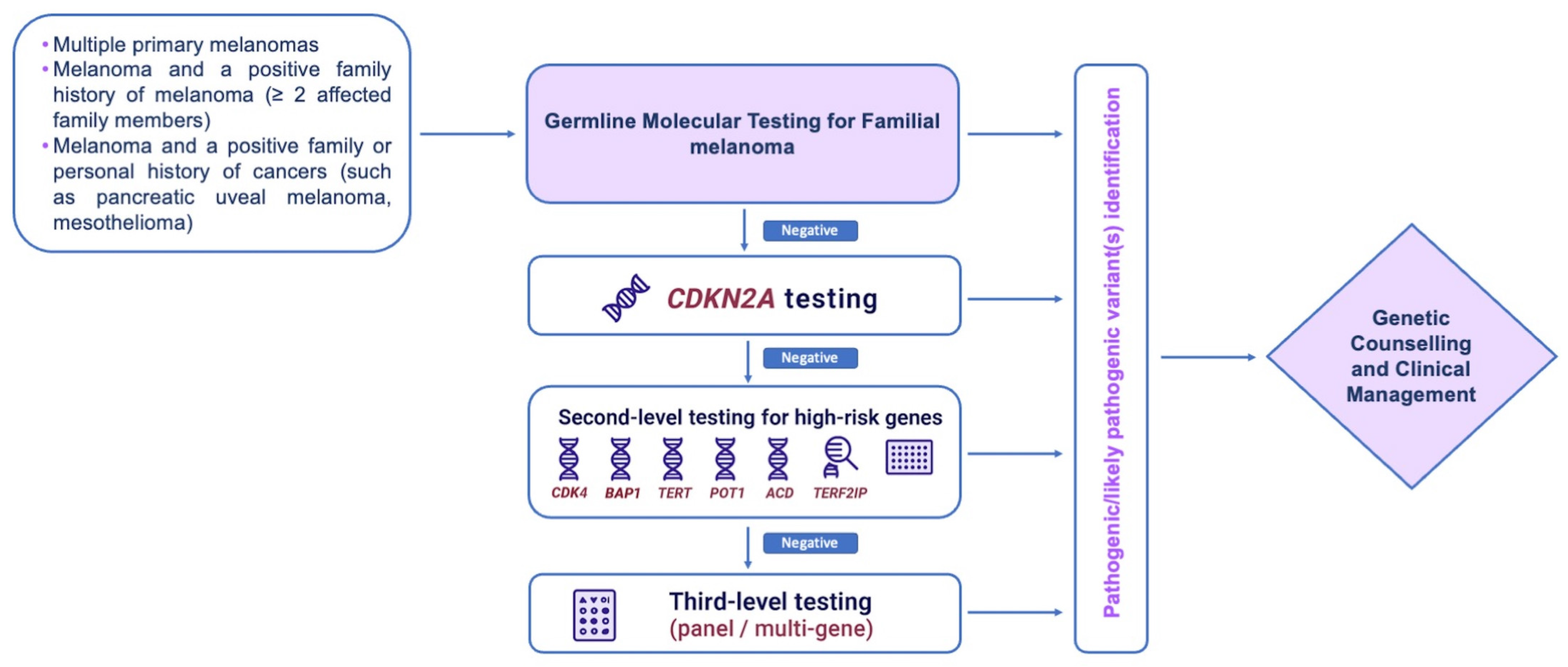
4. Genetic Testing
5. Genetic Counseling and Mutations Carriers’ Management
6. Role of Germline Variants in Prognosis and Therapy
7. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACD | ACD Shelterin Complex Subunit and Telomerase Recruitment Factor |
| AGR3 | Anterior Gradient 3 |
| ARNT | Aryl Hydrocarbon Receptor Nuclear Translocator |
| ASIP | Agouti Signaling Protein |
| BAP1 | BRCA1-Associated Protein 1 |
| BRAF | B-Raf Proto-Oncogene, Serine/Threonine Kinase |
| CCND1 | Cyclin D1 |
| CDK4 | Cyclin-Dependent Kinase 4 |
| CDKAL1 | CDK5 Regulatory Subunit-Associated Protein 1-Like 1 |
| CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A |
| CNV | Copy Number Variant |
| DC | Cyclin D–CDK4 Complex |
| FAMMM | Familial Atypical Multiple Mole Melanoma |
| GWAS | Genome-Wide Association Study |
| HERC2 | HECT and RLD Domain Containing E3 Ubiquitin Protein Ligase 2 |
| IL | Interleukin |
| IRF4 | Interferon Regulatory Factor 4 |
| MC1R | Melanocortin-1 Receptor |
| MGMT | O6-Methylguanine-DNA Methyltransferase |
| MITF | Melanocyte Inducing Transcription Factor |
| NGS | Next Generation Sequencing |
| OCA2 | Oculocutaneous Albinism II |
| OBFC1 | Oligosaccharide-Binding Fold Containing 1 |
| PARP1 | Poly (ADP-Ribose) Polymerase 1 |
| POT1 | Protection of Telomeres 1 |
| PTEN | Phosphatase and Tensin Homolog |
| RAD23B | RAD23 Homolog B |
| SH3PXD2A | SH3 and PX Domains 2A |
| SLC24A5 | Solute Carrier Family 24 Member 5 |
| SLC45A2 | Solute Carrier Family 45 Member 2 |
| SNP | Single Nucleotide Polymorphism |
| TERT | Telomerase Reverse Transcriptase |
| TERF2IP | TERF2 Interacting Protein |
| TPCN2 | Two-Pore Segment Channel 2 |
| TP53 | Tumor Protein P53 |
| TP53AIP1 | Tumor Protein P53 Regulated Apoptosis Inducing Protein 1 |
| TYR | Tyrosinase |
| TYRP1 | Tyrosinase Related Protein 1 |
| VDR | Vitamin D Receptor |
| VUS | Variant of Uncertain Significance |
| ZNF462 | Zinc Finger Protein 462 |
References
- Thomas, N.E.; Groben, P. Invasive superficial spreading melanomas arising from clinically normal skin. J. Am. Acad. Dermatol. 2004, 51, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Weatherhead, S.C.; Haniffa, M.; Lawrence, C.M. Melanomas Arising from Naevi and de Novo Melanomas—Does Origin Matter? Br. J. Dermatol. 2007, 156, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M.; Tucker, M.A. Dysplastic Nevi and Melanoma. Cancer Epidemiol. Biomark. Prev. 2013, 22, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Tschandl, P.; Berghoff, A.S.; Preusser, M.; Burgstaller-Muehlbacher, S.; Pehamberger, H.; Okamoto, I.; Kittler, H. NRAS and BRAF Mutations in Melanoma-Associated Nevi and Uninvolved Nevi. PLoS ONE 2013, 8, e0069639. [Google Scholar] [CrossRef] [PubMed]
- Shitara, D.; Tell-Martí, G.; Badenas, C.; Enokihara, M.M.S.S.; Alõs, L.; Larque, A.B.; Michalany, N.; Puig-Butille, J.A.; Carrera, C.; Malvehy, J.; et al. Mutational Status of Naevus-Associated Melanomas. Br. J. Dermatol. 2015, 173, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Cymerman, R.M.; Shao, Y.; Wang, K.; Zhang, Y.; Murzaku, E.C.; Penn, L.A.; Osman, I.; Polsky, D. De Novo vs Nevus-Associated Melanomas: Differences in Associations with Prognostic Indicators and Survival. J. Natl. Cancer Inst. 2016, 108, djw121. [Google Scholar] [CrossRef] [PubMed]
- Read, J.; Wadt, K.A.W.; Hayward, N.K. Melanoma Genetics. J. Med. Genet. 2016, 53, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Šerman, N.; Vranić, S.; Glibo, M.; Šerman, L.; Mokos, Z.B. Genetic Risk Factors in Melanoma Etiopathogenesis and the Role of Genetic Counseling: A Concise Review. Bosn. J. Basic Med. Sci. 2022, 22, 673–682. [Google Scholar] [PubMed]
- Precone, V.; Del Monaco, V.; Esposito, M.V.; De Palma, F.D.E.; Ruocco, A.; Salvatore, F.; D’Argenio, V. Cracking the Code of Human Diseases Using Next-Generation Sequencing: Applications, Challenges, and Perspectives. Biomed. Res. Int. 2015, 2015, 161648. [Google Scholar] [CrossRef] [PubMed]
- Scarano, C.; Veneruso, I.; De Simone, R.R.; Di Bonito, G.; Secondino, A.; D’Argenio, V. The Third-Generation Sequencing Challenge: Novel Insights for the Omic Sciences. Biomolecules 2024, 14, 568. [Google Scholar] [CrossRef] [PubMed]
- Nunziato, M.; Di Maggio, F.; Pensabene, M.; Esposito, M.V.; Starnone, F.; De Angelis, C.; Calabrese, A.; D’Aiuto, M.; Botti, G.; De Placido, S.; et al. Multi-gene panel testing increases germline predisposing mutations’ detection in a cohort of breast/ovarian cancer patients from Southern Italy. Front. Med. 2022, 9, 894358. [Google Scholar] [CrossRef] [PubMed]
- Secondino, A.; Starnone, F.; Veneruso, I.; Di Tella, M.A.; Conato, S.; De Angelis, C.; De Placido, S.; D’Argenio, V. Evaluation of a Four-Gene Panel for Hereditary Cancer Risk Assessment. Genes 2022, 13, 682. [Google Scholar] [CrossRef] [PubMed]
- Veneruso, I.; Ranieri, A.; Falcone, N.; Tripodi, L.; Scarano, C.; La Monica, I.; Pastore, L.; Lombardo, B.; D’Argenio, V. The Potential Usefulness of the Expanded Carrier Screening to Identify Hereditary Genetic Diseases: A Case Report from Real-World Data. Genes 2023, 14, 1651. [Google Scholar] [CrossRef] [PubMed]
- Fiasconaro, C.A.; Carbone, A.; Giordano, S.; Cavallo, F.; Fava, P.; Pasini, B.; Yakymiv, Y.; Marchisio, S.; Quaglino, P.; Ribero, S.; et al. Germline Non-CDKN2A Variants in Melanoma and Associated Hereditary Cancer Syndromes. Diseases 2025, 13, 180. [Google Scholar] [CrossRef] [PubMed]
- Leachman, S.A.; Carucci, J.; Kohlmann, W.; Banks, K.C.; Asgari, M.M.; Bergman, W.; Bianchi-Scarrà, G.; Brentnall, T.; Bressac-de Paillerets, B.; Bruno, W.; et al. Selection Criteria for Genetic Assessment of Patients with Familial Melanoma. J. Am. Acad. Dermatol. 2009, 61, 677.e1–677.e14. [Google Scholar] [CrossRef] [PubMed]
- Soura, E.; Eliades, P.J.; Shannon, K.; Stratigos, A.J.; Tsao, H. Hereditary Melanoma: Update on Syndromes and Management Emerging Melanoma Cancer Complexes and Genetic Counseling. J. Am. Acad. Dermatol. 2016, 74, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Soura, E.; Eliades, P.J.; Shannon, K.; Stratigos, A.J.; Tsao, H. Hereditary Melanoma: Update on Syndromes and Management Genetics of Familial Atypical Multiple Mole Melanoma Syndrome. J. Am. Acad. Dermatol. 2016, 74, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, K.J.; Jaju, P.D.; Tang, J.Y.; Carbone, M.; Leachman, S.; Sarin, K.Y. Familial Skin Cancer Syndromes: Increased Melanoma Risk. J. Am. Acad. Dermatol. 2016, 74, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.R.; El Moumni, M.; van Leeuwen, B.L.; van den Akker, P.C.; Rácz, E. Identifying high-risk melanoma patients: The importance of acquiring a detailed family history. J. Eur. Acad. Dermatol. Venereol. 2025, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Toussi, A.; Mans, N.; Welborn, J.; Kiuru, M. Germline Mutations Predisposing to Melanoma. J. Cutan. Pathol. 2020, 47, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Harland, M.; Cust, A.E.; Badenas, C.; Chang, Y.M.; Holland, E.A.; Aguilera, P.; Aitken, J.F.; Armstrong, B.K.; Barrett, J.H.; Carrera, C.; et al. Prevalence and Predictors of Germline CDKN2A Mutations for Melanoma Cases from Australia, Spain and the United Kingdom. Hered. Cancer Clin. Pract. 2014, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Hajkova, N.; Hojny, J.; Nemejcova, K.; Dundr, P.; Ulrych, J.; Jirsova, K.; Glezgova, J.; Ticha, I. Germline Mutation in the TP53 Gene in Uveal Melanoma. Sci. Rep. 2018, 8, 7618. [Google Scholar] [CrossRef] [PubMed]
- Leachman, S.A.; Lucero, O.M.; Sampson, J.E.; Cassidy, P.; Bruno, W.; Queirolo, P.; Ghiorzo, P. Identification, genetic testing, and management of hereditary melanoma. Cancer Metastasis Rev. 2017, 36, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Monnerat, C.; Chompret, A.; Kannengiesser, C.; Avril, M.F.; Janin, N.; Spatz, A.; Guinebretière, J.M.; Marian, C.; Barrois, M.; Boitier, F.; et al. BRCA1, BRCA2, TP53, and CDKN2A Germline Mutations in Patients with Breast Cancer and Cutaneous Melanoma. Fam. Cancer 2007, 6, 453–461. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Melanoma of the Skin-Cancer Stat Facts. National Cancer Institute. 2018. Available online: https://seer.cancer.gov/statfacts/html/melan.html (accessed on 15 May 2025).
- Potrony, M.; Badenas, C.; Aguilera, P.; Puig-Butille, J.A.; Carrera, C.; Malvehy, J.; Puig, S. Update in Genetic Susceptibility in Melanoma. Ann. Transl. Med. 2015, 3, 210. [Google Scholar] [PubMed]
- Funchain, P.; Ni, Y.; Heald, B.; Bungo, B.; Arbesman, M.; Behera, T.R.; McCormick, S.; Song, J.M.; Kennedy, L.B.; Nielsen, S.M.; et al. Germline Cancer Susceptibility in Individuals with Melanoma. J. Am. Acad. Dermatol. 2024, 91, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Potrony, M.; Puig-Butillé, J.A.; Aguilera, P.; Badenas, C.; Carrera, C.; Malvehy, J.; Puig, S. Increased Prevalence of Lung, Breast, and Pancreatic Cancers in Addition to Melanoma Risk in Families Bearing the Cyclin-Dependent Kinase Inhibitor 2A Mutation: Implications for Genetic Counseling. J. Am. Acad. Dermatol. 2014, 71, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, H.; Höiom, V.; Jönsson, G.; Tuominen, R.; Ingvar, C.; Borg, Å.; Olsson, H.; Hansson, J. High Risk of Tobacco-Related Cancers in CDKN2A Mutation-Positive Melanoma Families. J. Med. Genet. 2014, 51, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Gupta, S.; McCormick, S.R.; Tsao, H. New Insights into Melanoma Tumor Syndromes. JID Innov. Ski. Sci. Mol. Popul. Health 2022, 2, 100152. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M.; Chan, M.; Harland, M.; Gillanders, E.M.; Hayward, N.K.; Avril, M.F.; Azizi, E.; Bianchi-Scarra, G.; Bishop, D.T.; Bressac-De Paillerets, B.; et al. High-Risk Melanoma Susceptibility Genes and Pancreatic Cancer, Neural System Tumors, and Uveal Melanoma across GenoMEL. Cancer Res. 2006, 66, 9818–9828. [Google Scholar] [CrossRef] [PubMed]
- Pedace, L.; De Simone, P.; Castori, M.; Sperduti, I.; Silipo, V.; Eibenschutz, L.; De Bernardo, C.; Buccini, P.; Moscarella, E.; Panetta, C.; et al. Clinical Features Predicting Identification of CDKN2A Mutations in Italian Patients with Familial Cutaneous Melanoma. Cancer Epidemiol. 2011, 35, e116-20. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M.; Tucker, M.A. Genetic Epidemiology of Familial Melanoma. Dermatol. Clin. 1995, 3, 605–612. [Google Scholar] [CrossRef]
- Borg, Å.; Sandberg, T.; Nilsson, K.; Johannsson, O.; Klinker, M.; Måsbäck, A.; Westerdahl, J.; Olsson, H.; Ingvar, C. High Frequency of Multiple Melanomas and Breast and Pancreas Carcinomas in CDKN2A Mutation-Positive Melanoma Families. J. Natl. Cancer Inst. 2000, 92, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Cremin, C.; Howard, S.; Le, L.; Karsan, A.; Schaeffer, D.F.; Renouf, D.; Schrader, K.A. CDKN2A Founder Mutation in Pancreatic Ductal Adenocarcinoma Patients without Cutaneous Features of Familial Atypical Multiple Mole Melanoma (FAMMM) Syndrome. Hered. Cancer Clin. Pract. 2018, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, R.R.; Wieben, E.D.; Chaffee, K.G.; Antwi, S.O.; Raskin, L.; Olopade, O.I.; Li, D.; Highsmith, W.E.; Colon-Otero, G.; Khanna, L.G.; et al. CDKN2A Germline Rare Coding Variants and Risk of Pancreatic Cancer in Minority Populations. Cancer Epidemiol. Biomark. Prev. 2018, 27, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Garutti, M.; Targato, G.; Buriolla, S.; Palmero, L.; Minisini, A.M.; Puglisi, F. CDK4/6 Inhibitors in Melanoma: A Comprehensive Review. Cells 2021, 10, 1334. [Google Scholar] [CrossRef] [PubMed]
- Louie, B.H.; Kurzrock, R. BAP1: Not Just a BRCA1-Associated Protein. Cancer Treat. Rev. 2020, 90, 102091. [Google Scholar] [CrossRef] [PubMed]
- Masoomian, B.; Shields, J.A.; Shields, C.L. Overview of BAP1 Cancer Predisposition Syndrome and the Relationship to Uveal Melanoma. J. Curr. Ophthalmol. 2018, 30, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Wadt, K.A.; Aoude, L.G.; Johansson, P.; Solinas, A.; Pritchard, A.; Crainic, O.; Andersen, M.T.; Kiilgaard, J.F.; Heegaard, S.; Sunde, L.; et al. A Recurrent Germline BAP1 Mutation and Extension of the BAP1 Tumor Predisposition Spectrum to Include Basal Cell Carcinoma. Clin. Genet. 2015, 88, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Walpole, S.; Pritchard, A.L.; Cebulla, C.M.; Pilarski, R.; Stautberg, M.; Davidorf, F.H.; de la Fouchardière, A.; Cabaret, O.; Golmard, L.; Stoppa-Lyonnet, D.; et al. Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide. J. Natl. Cancer Inst. 2018, 110, 1328–1341. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, S.; Kong, H.; Srinivas, N.; Garcia-Casado, Z.; Requena, C.; Fallah, M.; Heidenreich, B.; Planelles, D.; Traves, V.; Schadendorf, D.; et al. Telomere Length, Telomerase Reverse Transcriptase Promoter Mutations, and Melanoma Risk. Genes Chromosomes Cancer 2018, 57, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chen, Y.; Zhang, L.; Ma, L.; Jiang, K.; Yao, G.; Zhu, L. TERT Promoter Mutations and Telomerase in Melanoma. J. Oncol. 2022, 2022, 6300329. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Bisht, K.; Savage, S.A.; Nandakumar, J.; Keegan, C.E.; Maillard, I. The Shelterin Complex and Hematopoiesis. J. Clin. Investig. 2016, 126, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.A. Beginning at the Ends: Telomeres and Human Disease. F1000Research 2018, 7, F1000. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Harland, M.; Petljak, M.; Robles-Espinoza, C.D.; Ding, Z.; Gruis, N.A.; van Doorn, R.; Pooley, K.A.; Dunning, A.M.; Aoude, L.G.; Wadt, K.A.W.; et al. Germline TERT Promoter Mutations Are Rare in Familial Melanoma. Fam. Cancer 2016, 15, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, A.; Meier, F.; Schlein, C.; Jansen, P.; Lodde, G.; Song, M.; Kretz, J.; Möller, I.; Stadtler, N.; Livingstone, E.; et al. Clinical and Pathological Characteristics of Familial Melanoma with Germline TERT Promoter Variants. Pigment. Cell Melanoma Res. 2022, 35, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Robles-Espinoza, C.D.; Harland, M.; Ramsay, A.J.; Aoude, L.G.; Quesada, V.; Ding, Z.; Pooley, K.A.; Pritchard, A.L.; Tiffen, J.C.; Petljak, M.; et al. POT1 Loss-of-Function Variants Predispose to Familial Melanoma. Nat. Genet. 2014, 46, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Yang, X.R.; Ballew, B.; Rotunno, M.; Calista, D.; Fargnoli, M.C.; Ghiorzo, P.; Bressac-De Paillerets, B.; Nagore, E.; Avril, M.F.; et al. Rare Missense Variants in POT1 Predispose to Familial Cutaneous Malignant Melanoma. Nat. Genet. 2014, 46, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.L.S.; Hattangady, N.; Lerario, A.M.; Williams, C.; Koeppe, E.; Quinonez, S.; Osborne, J.; Cha, K.B.; Else, T. A New POT1 Germline Mutation-Expanding the Spectrum of POT1-Associated Cancers. Fam. Cancer 2017, 16, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Krunic, M.; Wendt, J.; von Haeseler, A.; Okamoto, I. Germline Variants in the POT1-Gene in High-Risk Melanoma Patients in Austria. G3 Bethesda 2018, 8, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- Potrony, M.; Puig-Butille, J.A.; Ribera-Sola, M.; Iyer, V.; Robles-Espinoza, C.D.; Aguilera, P.; Carrera, C.; Malvehy, J.; Badenas, C.; Landi, M.T.; et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br. J. Dermatol. 2019, 181, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Robles-Espinoza, C.D.; Rodriguez, D.; Rudat, S.S.; Puig, S.; Potrony, M.; Wong, C.C.; Hewinson, J.; Aguilera, P.; Puig-Butille, J.A.; et al. Association of the POT1 Germline Missense Variant p.I78T with Familial Melanoma. JAMA Dermatol. 2019, 155, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Abu Shtaya, A.; Kedar, I.; Bazak, L.; Basel-Salmon, L.; Barhom, S.F.; Naftali, M.; Eskin-Schwartz, M.; Birk, O.S.; Polager-Modan, S.; Keidar, N.; et al. A POT1 Founder Variant Associated with Early Onset Recurrent Melanoma and Various Solid Malignancies. Genes 2024, 15, 355. [Google Scholar] [CrossRef] [PubMed]
- Aoude, L.G.; Pritchard, A.L.; Robles-Espinoza, C.D.; Wadt, K.; Harland, M.; Choi, J.; Gartside, M.; Quesada, V.; Johansson, P.; Palmer, J.M.; et al. Nonsense Mutations in the Shelterin Complex Genes ACD and TERF2IP in Familial Melanoma. J. Natl. Cancer Inst. 2014, 107, dju408. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M.; Qin, R.; Chu, E.Y.; Elder, D.E.; Massi, D.; Adams, D.J.; Harms, P.W.; Robles-Espinoza, C.D.; Newton-Bishop, J.A.; Bishop, D.T.; et al. Association of Germline Variants in Telomere Maintenance Genes (POT1, TERF2IP, ACD, and TERT) with Spitzoid Morphology in Familial Melanoma: A Multi-Center Case Series. JAAD Int. 2023, 11, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, E.; Fargnoli, M.C.; Gandini, S.; Maisonneuve, P.; Liu, F.; Kayser, M.; Nijsten, T.; Han, J.; Kumar, R.; Gruis, N.A.; et al. MC1R Gene Variants and Non-Melanoma Skin Cancer: A Pooled-Analysis from the M-SKIP Project. Br. J. Cancer 2015, 113, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Wendt, J.; Mueller, C.; Rauscher, S.; Fae, I.; Fischer, G.; Okamoto, I. Contributions by MC1R Variants to Melanoma Risk in Males and Females. JAMA Dermatol. 2018, 154, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Potjer, T.P.; Bollen, S.; Grimbergen, A.J.E.M.; van Doorn, R.; Gruis, N.A.; van Asperen, C.J.; Hes, F.J.; van der Stoep, N. Multigene Panel Sequencing of Established and Candidate Melanoma Susceptibility Genes in a Large Cohort of Dutch Non-CDKN2A/CDK4 Melanoma Families. Int. J. Cancer 2019, 144, 2453–2464. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Czyz, M. MITF in Melanoma: Mechanisms behind Its Expression and Activity. Cell. Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Ciccarese, G.; Dalmasso, B.; Bruno, W.; Queirolo, P.; Pastorino, L.; Andreotti, V.; Spagnolo, F.; Tanda, E.; Ponti, G.; Massone, C.; et al. Clinical, pathological and dermoscopic phenotype of MITF p.E318K carrier cutaneous melanoma patients. J. Transl. Med. 2020, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Woods, S.L.; Boyle, G.M.; Aoude, L.G.; MacGregor, S.; Zismann, V.; Gartside, M.; Cust, A.E.; Haq, R.; Harland, M.; et al. A Novel Recurrent Mutation in MITF Predisposes to Familial and Sporadic Melanoma. Nature 2011, 480, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Potrony, M.; Puig-Butille, J.A.; Aguilera, P.; Badenas, C.; Tell-Marti, G.; Carrera, C.; Del Pozo, L.J.; Conejo-Mir, J.; Malvehy, J.; Puig, S. Prevalence of MITF p.E318K in Patients with Melanoma Independent of the Presence of CDKN2A Causative Mutations. JAMA Dermatol. 2016, 152, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M.; Xiao, Y.; Sampson, J.; Zhu, B.; Rotunno, M.; Bennett, H.; Wen, Y.; Jones, K.; Vogt, A.; Burdette, L.; et al. Rare Germline Variants in Known Melanoma Susceptibility Genes in Familial Melanoma. Hum. Mol. Genet. 2017, 26, 4886–4895. [Google Scholar] [CrossRef] [PubMed]
- Appelqvist, F.; Yhr, M.; Erlandson, A.; Martinsson, T.; Enerbäck, C. Deletion of the MGMT Gene in Familial Melanoma. Genes Chromosomes Cancer 2014, 53, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Yin, Z.; Wang, G.; Zeng, S. Clinical and Prognostic Significance of O6-Methylguanine-DNA Methyltransferase Promoter Methylation in Patients with Melanoma: A Systematic Meta-Analysis. Ann. Dermatol. 2018, 30, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Helsing, P.; Nymoen, D.A.; Rootwelt, H.; Vårdal, M.; Akslen, L.A.; Molven, A.; Andresen, P.A. MC1R, ASIP, TYR, and TYRP1 gene variants in a population-based series of multiple primary melanomas. Genes Chromosomes Cancer 2012, 51, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Pho, L.N.; Leachman, S.A. Genetics of Pigmentation and Melanoma Predisposition. G. Ital. Dermatol. Venereol. 2010, 1, 37–45. [Google Scholar]
- Gibbs, D.C.; Small, B.M.; Autuori, I.; Leong, S.F.; Ali, E.; Kenney, J.; Luo, L.; Kanetsky, P.A.; Busam, K.J.; Cust, A.E.; et al. Association of Inherited Genetic Variants with Multiple Primary Melanoma. Cancer Epidemiol. Biomark. Prev. 2025, 34, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Kocarnik, J.M.; Park, S.L.; Han, J.; Dumitrescu, L.; Cheng, I.; Wilkens, L.R.; Schumacher, F.R.; Kolonel, L.; Carlson, C.S.; Crawford, D.C.; et al. Pleiotropic and Sex-Specific Effects of Cancer GWAS SNPs on Melanoma Risk in the Population Architecture Using Genomics and Epidemiology (PAGE) Study. PLoS ONE 2015, 10, e.0120491. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, L.; Rachakonda, P.S.; Scherer, D.; Bermejo, J.L.; Planelles, D.; Requena, C.; Hemminki, K.; Nagore, E.; Kumar, R. Variants at Chromosome 20 (ASIP Locus) and Melanoma Risk. Int. J. Cancer 2013, 132, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Nan, H.; Kraft, P.; Hunter, D.J.; Han, J. Genetic Variants in Pigmentation Genes, Pigmentary Phenotypes, and Risk of Skin Cancer in Caucasians. Int. J. Cancer 2009, 125, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Song, F.; Liang, L.; Nan, H.; Zhang, J.; Liu, H.; Wang, L.E.; Wei, Q.; Lee, J.E.; Amos, C.I.; et al. Genome-Wide Association Studies Identify Several New Loci Associated with Pigmentation Traits and Skin Cancer Risk in European Americans. Hum. Mol. Genet. 2013, 22, 2948–2959. [Google Scholar] [CrossRef] [PubMed]
- Nathan, V.; Johansson, P.A.; Palmer, J.M.; Howlie, M.; Hamilton, H.R.; Wadt, K.; Jönsson, G.; Brooks, K.M.; Pritchard, A.L.; Hayward, N.K. Germline Variants in Oculocutaneous Albinism Genes and Predisposition to Familial Cutaneous Melanoma. Pigment. Cell Melanoma Res. 2019, 32, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Law, M.H.; Bishop, D.T.; Lee, J.E.; Brossard, M.; Martin, N.G.; Moses, E.K.; Song, F.; Barrett, J.H.; Kumar, R.; Easton, D.F.; et al. Genome-Wide Meta-Analysis Identifies Five New Susceptibility Loci for Cutaneous Malignant Melanoma. Nat. Genet. 2015, 47, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, K.; Martin-Hirsch, P.L.; Martin, F.L. CYP1B1 and Hormone-Induced Cancer. Cancer Lett. 2012, 324, 13–30. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, S.; Montgomery, G.W.; Liu, J.Z.; Zhao, Z.Z.; Henders, A.K.; Stark, M.; Schmid, H.; Holland, E.A.; Duffy, D.L.; Zhang, M.; et al. Genome-Wide Association Study Identifies a New Melanoma Susceptibility Locus at 1q21.3. Nat. Genet. 2011, 43, 1114–1118. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kraft, P.; Nan, H.; Guo, Q.; Chen, C.; Qureshi, A.; Hankinson, S.E.; Hu, F.B.; Duffy, D.L.; Zhen, Z.Z.; et al. A Genome-Wide Association Study Identifies Novel Alleles Associated with Hair Color and Skin Pigmentation. PLoS Genet. 2008, 4, e1000074. [Google Scholar] [CrossRef] [PubMed]
- Duffy, D.L.; Iles, M.M.; Glass, D.; Zhu, G.; Barrett, J.H.; Höiom, V.; Zhao, Z.Z.; Sturm, R.A.; Soranzo, N.; Hammond, C.; et al. IRF4 Variants Have Age-Specific Effects on Nevus Count and Predispose to Melanoma. Am. J. Hum. Genet. 2010, 87, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, G.C.; Patel, S.; Tyson, K.; Adam, P.J.; Schenker, M.; Loader, J.A.; Daviet, L.; Legrain, P.; Parekh, R.; Harris, A.L.; et al. HAG-2 and HAG-3, Human Homologues of Genes Involved in Differentiation, Are Associated with Oestrogen Receptor-Positive Breast Tumours and Interact with Metastasis Gene C4.4a and Dystroglycan. Br. J. Cancer 2003, 88, 579–585. [Google Scholar] [CrossRef] [PubMed]
- King, E.R.; Tung, C.S.; Tsang, Y.T.M.; Zu, Z.; Lok, G.T.M.; Deavers, M.T.; Malpica, A.; Wolf, J.K.; Lu, K.H.; Birrer, M.J.; et al. The Anterior Gradient Homolog 3 (AGR3) Gene Is Associated with Differentiation and Survival in Ovarian Cancer. Am. J. Surg. Pathol. 2011, 35, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, B. Immunology of Melanoma. Clin. Dermatol. 2013, 31, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune System and Melanoma Biology: A Balance between Immunosurveillance and Immune Escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef] [PubMed]
- Marzagalli, M.; Ebelt, N.D.; Manuel, E.R. Unraveling the Crosstalk between Melanoma and Immune Cells in the Tumor Microenvironment. Semin. Cancer Biol. 2019, 59, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Eddy, K.; Chen, S. Overcoming Immune Evasion in Melanoma. Int. J. Mol. Sci. 2020, 21, 8984. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, C.C.; Huff, C.; Stevens, J.; Yu, Y.; Holmen, S.L.; Silvis, M.R.; Trombetti, K.; Zhao, H.; Grossman, D.; Farnham, J.M.; et al. A Nonsynonymous Variant in the GOLM1 Gene in Cutaneous Malignant Melanoma. J. Natl. Cancer Inst. 2018, 110, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Benfodda, M.; Gazal, S.; Descamps, V.; Basset-Seguin, N.; Deschamps, L.; Thomas, L.; Lebbe, C.; Saiag, P.; Zanetti, R.; Sacchetto, L.; et al. Truncating Mutations of TP53AIP1 Gene Predispose to Cutaneous Melanoma. Genes Chromosomes Cancer 2018, 57, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Gillanders, E.; Juo, S.H.H.; Holland, E.A.; Jones, M.P.; Nancarrow, D.; Freas-Lutz, D.; Sood, R.; Park, N.; Faruque, M.; Markey, C.; et al. Localization of a Novel Melanoma Susceptibility Locus to 1p22. Am. J. Hum. Genet. 2003, 73, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Höiom, V.; Tuominen, R.; Hansson, J. Genome-Wide Linkage Analysis of Swedish Families to Identify Putative Susceptibility Loci for Cutaneous Malignant Melanoma. Genes Chromosomes Cancer 2011, 50, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Cannon-Albright, L.A.; Teerlink, C.C.; Farnham, J.M.; Thomas, A.W.; Zone, J.J.; Leachman, S.A. Linkage Analysis of Extended High-Risk Pedigrees Replicates a Cutaneous Malignant Melanoma Predisposition Locus on Chromosome 9q21. J. Investig. Dermatol. 2013, 133, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Tuominen, R.; Jönsson, G.; Enerbäck, C.; Appelqvist, F.; Olsson, H.; Ingvar, C.; Hansson, J.; Höiom, V. Investigation of a Putative Melanoma Susceptibility Locus at Chromosome 3q29. Cancer Genet. 2014, 207, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Potrony, M.; Puig-Butille, J.A.; Farnham, J.M.; Giménez-Xavier, P.; Badenas, C.; Tell-Martí, G.; Aguilera, P.; Carrera, C.; Malvehy, J.; Teerlink, C.C.; et al. Genome-Wide Linkage Analysis in Spanish Melanoma-Prone Families Identifies a New Familial Melanoma Susceptibility Locus at 11q. Eur. J. Hum. Genet. 2018, 26, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.T.; Bishop, D.T.; MacGregor, S.; Machiela, M.J.; Stratigos, A.J.; Ghiorzo, P.; Brossard, M.; Calista, D.; Choi, J.; Fargnoli, M.C.; et al. Genome-Wide Association Meta-Analyses Combining Multiple Risk Phenotypes Provide Insights into the Genetic Architecture of Cutaneous Melanoma Susceptibility. Nat. Genet. 2020, 52, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Pellegrini, C.; Cardelli, L.; Ciciarelli, V.; Nardo, L.D.; Fargnoli, M.C. Familial Melanoma: Diagnostic and Management Implications. Dermatol. Pract. Concept. 2019, 9, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Cirino, A.L.; Lakdawala, N.K.; McDonough, B.; Conner, L.; Adler, D.; Weinfeld, M.; O’Gara, P.; Rehm, H.L.; MacHini, K.; Lebo, M.; et al. A Comparison of Whole Genome Sequencing to Multigene Panel Testing in Hypertrophic Cardiomyopathy Patients. Circ. Cardiovasc. Genet. 2017, 10, e.001768. [Google Scholar] [CrossRef] [PubMed]
- Kuo, F.C.; Mar, B.G.; Lindsley, R.C.; Lindeman, N.I. The Relative Utilities of Genome-Wide, Gene Panel, and Individual Gene Sequencing in Clinical Practice. Blood 2017, 130, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Mazzaccara, C.; Lombardi, R.; Mirra, B.; Barretta, F.; Esposito, M.V.; Uomo, F.; Caiazza, M.; Monda, E.; Losi, M.A.; Limongelli, G.; et al. Next-Generation Sequencing Gene Panels in Inheritable Cardiomyopathies and Channelopathies: Prevalence of Pathogenic Variants and Variants of Unknown Significance in Uncommon Genes. Biomolecules 2022, 12, 1417. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, A.; Veneruso, I.; La Monica, I.; Pascale, M.G.; Pastore, L.; D’argenio, V.; Lombardo, B. Combined ACGH and Exome Sequencing Analysis Improves Autism Spectrum Disorders Diagnosis: A Case Report. Medicina 2022, 58, 522. [Google Scholar] [CrossRef] [PubMed]
- Desmond, A.; Kurian, A.W.; Gabree, M.; Mills, M.A.; Anderson, M.J.; Kobayashi, Y.; Horick, N.; Yang, S.; Shannon, K.M.; Tung, N.; et al. Clinical Actionability of Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Risk Assessment. JAMA Oncol. 2015, 1, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Neben, C.L.; Zimmer, A.D.; Stedden, W.; van den Akker, J.; O’Connor, R.; Chan, R.C.; Chen, E.; Tan, Z.; Leon, A.; Ji, J.; et al. Multi-Gene Panel Testing of 23,179 Individuals for Hereditary Cancer Risk Identifies Pathogenic Variant Carriers Missed by Current Genetic Testing Guidelines. J. Mol. Diagn. 2019, 21, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Veneruso, I.; Scarano, C.; D’Argenio, V. Hereditary Breast Cancer Susceptibility Genes. Biochim. Clin. 2023, 47, 328–339. [Google Scholar]
- Taber, J.M.; Aspinwall, L.G.; Stump, T.K.; Kohlmann, W.; Champine, M.; Leachman, S.A. Genetic Test Reporting Enhances Understanding of Risk Information and Acceptance of Prevention Recommendations Compared to Family History-Based Counseling Alone. J. Behav. Med. 2015, 38, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Bruno, W.; Dalmasso, B.; Barile, M.; Andreotti, V.; Elefanti, L.; Colombino, M.; Vanni, I.; Allavena, E.; Barbero, F.; Passoni, E.; et al. Predictors of Germline Status for Hereditary Melanoma: 5 Years of Multi-Gene Panel Testing within the Italian Melanoma Intergroup. ESMO Open 2022, 7, 100525. [Google Scholar] [CrossRef] [PubMed]
- Jones, O.T.; Jurascheck, L.C.; Van Melle, M.A.; Hickman, S.; Burrows, N.P.; Hall, P.N.; Emery, J.; Walter, F.M. Dermoscopy for Melanoma Detection and Triage in Primary Care: A Systematic Review. BMJ Open 2019, 9, e027529. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M.; Struewing, J.P.; Chidambaram, A.; Fraser, M.C.; Tucker, M.A. Genotype-Phenotype Relationships in U.S. Melanoma-Prone Families with CDKN2A and CDK4 Mutations. J. Natl. Cancer Inst. 2000, 92, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Kefford, R.F.; Newton Bishop, J.A.; Bergman, W.; Tucker, M.A. Counseling and DNA Testing for Individuals Perceived to Be Genetically Predisposed to Melanoma: A Consensus Statement of the Melanoma Genetics Consortium. J. Clin. Oncol. 1999, 17, 3245–3251. [Google Scholar] [CrossRef] [PubMed]
- Haenssle, H.A.; Korpas, B.; Hansen-Hagge, C.; Buhl, T.; Kaune, K.M.; Johnsen, S.; Rosenberger, A.; Schön, M.P.; Emmert, S. Selection of Patients for Long-Term Surveillance with Digital Dermoscopy by Assessment of Melanoma Risk Factors. Arch. Dermatol. 2010, 146, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Salerni, G.; Carrera, C.; Lovatto, L.; Puig-Butille, J.A.; Badenas, C.; Plana, E.; Puig, S.; Malvehy, J. Benefits of Total Body Photography and Digital Dermatoscopy (“two-Step Method of Digital Follow-up”) in the Early Diagnosis of Melanoma in Patients at High Risk for Melanoma. J. Am. Acad. Dermatol. 2012, 67, e17–e27. [Google Scholar] [CrossRef] [PubMed]
- Moloney, F.J.; Guitera, P.; Coates, E.; Haass, N.K.; Ho, K.; Khoury, R.; O’Connell, R.L.; Raudonikis, L.; Schmid, H.; Mann, G.J.; et al. Detection of Primary Melanoma in Individuals at Extreme High Risk: A Prospective 5-Year Follow-up Study. JAMA Dermatol. 2014, 150, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Rademaker, M.; Oakley, A. Digital Monitoring by Whole Body Photography and Sequential Digital Dermoscopy Detects Thinner Melanomas. J. Prim. Health Care 2010, 4, 268–272. [Google Scholar] [CrossRef]
- Watts, C.G.; Cust, A.E.; Menzies, S.W.; Coates, E.; Mann, G.J.; Morton, R.L. Specialized Surveillance for Individuals at High Risk for Melanoma: A Cost Analysis of a High-Risk Clinic. JAMA Dermatol. 2015, 151, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.R.; Chang, Y.M.; Bishop, D.T.; Armstrong, B.K.; Bataille, V.; Bergman, W.; Berwick, M.; Bracci, P.M.; Elwood, J.M.; Ernstoff, M.S.; et al. Development and Validation of a Melanoma Risk Score Based on Pooled Data from 16 Case-Control Studies. Cancer Epidemiol. Biomark. Prev. 2015, 24, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.G.; Ransohoff, K.J.; Yang, L.; Hedlin, H.; Assimes, T.; Han, J.; Stefanick, M.; Tang, J.Y.; Sarin, K.Y. Melanoma Risk Prediction Using a Multilocus Genetic Risk Score in the Women’s Health Initiative Cohort. J. Am. Acad. Dermatol. 2018, 79, 36–41.e10. [Google Scholar] [CrossRef] [PubMed]
- Cust, A.E.; Drummond, M.; Kanetsky, P.A.; Mann, G.J.; Schmid, H.; Hopper, J.L.; Aitken, J.F.; Armstrong, B.K.; Giles, G.G.; Holland, E.; et al. Assessing the Incremental Contribution of Common Genomic Variants to Melanoma Risk Prediction in Two Population-Based Studies. J. Investig. Dermatol. 2018, 138, 2617–2624. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Chen, T.H.; Pfeiffer, R.M.; Fargnoli, M.C.; Calista, D.; Ghiorzo, P.; Peris, K.; Puig, S.; Menin, C.; De Nicolo, A.; et al. Combining Common Genetic Variants and Non-Genetic Risk Factors to Predict Risk of Cutaneous Melanoma. Hum. Mol. Genet. 2018, 27, 4145–4156. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.R.; Randerson-Moor, J.; Kukalizch, K.; Harland, M.; Kumar, R.; Madhusudan, S.; Nagore, E.; Hansson, J.; Höiom, V.; Ghiorzo, P.; et al. Inherited Variants in the MC1R Gene and Survival from Cutaneous Melanoma: A BioGenoMEL Study. Pigment. Cell Melanoma Res. 2012, 25, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.J.; Reiner, A.S.; Begg, C.B.; Cust, A.E.; Busam, K.J.; Anton-Culver, H.; Dwyer, T.; From, L.; Gallagher, R.P.; Gruber, S.B.; et al. Inherited Variation at MC1R and ASIP and Association with Melanoma-Specific Survival. Int. J. Cancer 2015, 136, 2659–2667. [Google Scholar] [CrossRef] [PubMed]
- Lira, F.E.; Podlipnik, S.; Potrony, M.; Tell-Martí, G.; Calbet-Llopart, N.; Barreiro, A.; Carrera, C.; Malvehy, J.; Puig, S. Inherited MC1R Variants in Patients with Melanoma Are Associated with Better Survival in Women. Br. J. Dermatol. 2020, 182, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.T.; Kanetsky, P.A.; Tsang, S.; Gold, B.; Munroe, D.; Rebbeck, T.; Swoyer, J.; Ter-Minassian, M.; Hedayati, M.; Grossman, L.; et al. MC1R, ASIP, and DNA Repair in Sporadic and Familial Melanoma in a Mediterranean Population. J. Natl. Cancer Inst. 2005, 97, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.R.; Jewell, R.; Affleck, P.; Anic, G.M.; Randerson-Moor, J.; Ozola, A.; Egan, K.M.; Elliott, F.; García-Casado, Z.; Hansson, J.; et al. Inherited Variation in the PARP1 Gene and Survival from Melanoma. Int. J. Cancer 2014, 135, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Bilinski, K.; Boyages, J. Association between 25-Hydroxyvitamin D Concentration and Breast Cancer Risk in an Australian Population: An Observational Case-Control Study. Breast Cancer Res. Treat. 2013, 137, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Ordonez-Mena, J.M.; Schottker, B.; Haug, U.; Muller, H.; Kohrle, J.; Schomburg, L.; Holleczek, B.; Hermann, B.H. Serum 25-Hydroxyvitamin d and Cancer Risk in Older Adults: Results from a Large German Prospective Cohort Study. Cancer Epidemiol. Biomark. Prev. 2013, 22, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Orlow, I.; Reiner, A.S.; Thomas, N.E.; Roy, P.; Kanetsky, P.A.; Luo, L.; Paine, S.; Armstrong, B.K.; Kricker, A.; Marrett, L.D.; et al. Vitamin D Receptor Polymorphisms and Survival in Patients with Cutaneous Melanoma: A Population-Based Study. Carcinogenesis 2016, 37, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Sundram, U.; Harvell, J.D.; Rouse, R.V.; Natkunam, Y. Expression of the B-Cell Proliferation Marker MUM1 by Melanocytic Lesions and Comparison with S100, Gp100 (HMB45), and MelanA. Mod. Pathol. 2003, 16, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Peña-Chilet, M.; Blanquer-Maceiras, M.; Ibarrola-Villava, M.; Martinez-Cadenas, C.; Martin-Gonzalez, M.; Gomez-Fernandez, C.; Mayor, M.; Aviles, J.A.; Lluch, A.; Ribas, G. Genetic Variants in PARP1 (Rs3219090) and IRF4 (Rs12203592) Genes Associated with Melanoma Susceptibility in a Spanish Population. BMC Cancer 2013, 13, 160. [Google Scholar] [CrossRef] [PubMed]
- Potrony, M.; Rebollo-Morell, A.; Giménez-Xavier, P.; Zimmer, L.; Puig-Butille, J.A.; Tell-Marti, G.; Sucker, A.; Badenas, C.; Carrera, C.; Malvehy, J.; et al. IRF4 Rs12203592 Functional Variant and Melanoma Survival. Int. J. Cancer 2017, 140, 1845–1849. [Google Scholar] [CrossRef] [PubMed]
- Florell, S.R.; Boucher, K.M.; Garibotti, G.; Astle, J.; Kerber, R.; Mineau, G.; Wiggins, C.; Noyes, R.D.; Tsodikov, A.; Cannon-Albright, L.A.; et al. Population-Based Analysis of Prognostic Factors and Survival in Familial Melanoma. J. Clin. Oncol. 2005, 23, 7168–7177. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, V.; Savarese, I.; D’Errico, A.; Gori, A.; Papi, F.; Colombino, M.; Sini, M.C.; Grazzini, M.; Stanganelli, I.; Rossari, S.; et al. Epidemiological Features and Prognostic Parameters of Multiple Primary Melanomas in CDKN2A-Mutations Patients. Pigment. Cell Melanoma Res. 2015, 28, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, H.; Hoiom, V.; Tuominen, R.; Nielsen, K.; Jonsson, G.; Olsson, H.; Hansson, J. Germline CDKN2A Mutation Status and Survival in Familial Melanoma Cases. J. Natl. Cancer Inst. 2016, 108, 11. [Google Scholar] [CrossRef] [PubMed]
- Dalmasso, B.; Pastorino, L.; Ciccarese, G.; Andreotti, V.; Grillo, F.; Mastracci, L.; Spagnolo, F.; Ballestrero, A.; Queirolo, P.; Bruno, W.; et al. CDKN2A Germline Mutations Are Not Associated with Poor Survival in an Italian Cohort of Melanoma Patients. J. Am. Acad. Dermatol. 2019, 80, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Domingues, B.; Lopes, J.M.; Soares, P.; Pópulo, H. Melanoma Treatment in Review. Immunotargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wang, H.; Li, C. Signal Pathways of Melanoma and Targeted Therapy. Signal Transduct. Target. Ther. 2021, 6, 424. [Google Scholar] [CrossRef] [PubMed]
- Kreuger, I.Z.M.; Slieker, R.C.; van Groningen, T.; van Doorn, R. Therapeutic Strategies for Targeting CDKN2A Loss in Melanoma. J. Investig. Dermatol. 2023, 143, 18–25.e1. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, P.F.; Lens, M.; Cocorocchio, E. Combined BRAF-Targeted Therapy with Immunotherapy in BRAF-Mutated Advanced Melanoma Patients. Curr. Oncol. Rep. 2021, 23, 138. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Schalper, K.; Sosman, J. Targeted Therapy and Immunotherapy: Emerging Biomarkers in Metastatic Melanoma. Pigment. Cell Melanoma Res. 2020, 33, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Castet, F.; Garcia-Mulero, S.; Sanz-Pamplona, R.; Cuellar, A.; Casanovas, O.; Caminal, J.M.; Piulats, J.M. Uveal Melanoma, Angiogenesis and Immunotherapy, Is There Any Hope? Cancers 2019, 11, 834. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Arya, A.; Iams, W.; Cruz, M.R.; Chandra, S.; Choi, J.; Giles, F. Current Landscape and Future of Dual Anti-CTLA4 and PD-1/PD-L1 Blockade Immunotherapy in Cancer; Lessons Learned from Clinical Trials with Melanoma and Non-Small Cell Lung Cancer (NSCLC). J. Immunother. Cancer 2018, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; Maclennan, G.T.; Montironi, R. Molecular Testing for BRAF Mutations to Inform Melanoma Treatment Decisions: A Move toward Precision Medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, K.; Briand, C.; Bellutti, F.; Schicher, N.; Blunder, S.; Zojer, M.; Hoeller, C. The Interplay of CDK4 and CDK6 in Melanoma. Oncotarget 2019, 10, 1346–1359. [Google Scholar] [CrossRef] [PubMed]
- Lugowska, I.; Teterycz, P.; Rutkowski, P. Immunotherapy of Melanoma. Contemp. Oncol. 2018, 22, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wu, Y.; Zhang, T.; Song, H.; Jv, H.; Guo, W.; Ren, G. The Clinical Significance of C-Kit Mutations in Metastatic Oral Mucosal Melanoma in China. Oncotarget 2017, 8, 82661–82673. [Google Scholar] [CrossRef] [PubMed]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A Moving Target in Immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.A.; Cheng, M.Y.; Mitra, A.; Ogawa, H.; Shi, V.Y.; Olney, L.P.; Kloxin, A.M.; Maverakis, E. MEK Inhibitors and Their Potential in the Treatment of Advanced Melanoma: The Advantages of Combination Therapy. Drug Des. Devel Ther. 2015, 10, 43–52. [Google Scholar] [PubMed]
- Liu, H.; Gou, X.; Tan, Y.; Fan, Q.; Chen, J. Immunotherapy and Delivery Systems for Melanoma. Hum. Vaccin. Immunother. 2024, 20, 2394252. [Google Scholar] [CrossRef] [PubMed]
- Amarillo, D.; Flaherty, K.T.; Sullivan, R.J. Targeted Therapy Innovations for Melanoma. Hematol. Oncol. Clin. North Am. 2024, 38, 973–995. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, H.; Olsson, H.; Tucker, M.A.; Yang, X.R.; Höiom, V.; Goldstein, A.M. Phenocopies in Melanoma-Prone Families with Germ-Line CDKN2A Mutations. Genet. Med. 2018, 20, 1087–1090. [Google Scholar] [CrossRef] [PubMed]
- Guida, S.; Ciardo, S.; De Pace, B.; De Carvalho, N.; Peccerillo, F.; Manfredini, M.; Farnetani, F.; Chester, J.; Kaleci, S.; Manganelli, M.; et al. The Influence of MC1R on Dermal Morphological Features of Photo-Exposed Skin in Women Revealed by Reflectance Confocal Microscopy and Optical Coherence Tomography. Exp. Dermatol. 2019, 28, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Chat, V.; Ferguson, R.; Simpson, D.; Kazlow, E.; Lax, R.; Moran, U.; Pavlick, A.; Frederick, D.; Boland, G.; Sullivan, R.; et al. Autoimmune Genetic Risk Variants as Germline Biomarkers of Response to Melanoma Immune-Checkpoint Inhibition. Cancer Immunol. Immunother. 2019, 68, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Meyle, K.D.; Guldberg, P. Genetic Risk Factors for Melanoma. Hum. Genet. 2009, 126, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Marzuka-Alcalá, A.; Gabree, M.J.; Tsao, H. Melanoma Susceptibility Genes and Risk Assessment. Methods Mol. Biol. 2014, 1102, 381–393. [Google Scholar] [PubMed]
- D’Argenio, V. The High-Throughput Analyses Era: Are We Ready for the Data Struggle? High Throughput 2018, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhou, W.; Zhu, B.; Hyland, P.L.; Bennett, H.; Xiao, Y.; Zhang, X.; Burke, L.S.; Song, L.; Hsu, C.H.; et al. Rare Germline Copy Number Variations and Disease Susceptibility in Familial Melanoma. J. Investig. Dermatol. 2016, 136, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.R.; Brown, K.; Landi, M.T.; Ghiorzo, P.; Badenas, C.; Xu, M.; Hayward, N.K.; Calista, D.; Landi, G.; Bruno, W.; et al. Duplication of CXC Chemokine Genes on Chromosome 4q13 in a Melanoma-Prone Family. Pigment. Cell Melanoma Res. 2012, 25, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Rocca, M.S.; Benna, C.; Mocellin, S.; Rossi, C.R.; Msaki, A.; Di Nisio, A.; Opocher, G.; Foresta, C. E2F1 Germline Copy Number Variations and Melanoma Susceptibility. J. Transl. Med. 2019, 17, 181. [Google Scholar] [CrossRef] [PubMed]
- Iyevleva, A.G.; Imyanitov, E.N. Cytotoxic and Targeted Therapy for Hereditary Cancers. Hered. Cancer Clin. Pract. 2016, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Lima, Z.S.; Ghadamzadeh, M.; Arashloo, F.T.; Amjad, G.; Ebadi, M.R.; Younesi, L. Recent Advances of Therapeutic Targets Based on the Molecular Signature in Breast Cancer: Genetic Mutations and Implications for Current Treatment Paradigms. J. Hematol. Oncol. 2019, 12, 38. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Gene | Name | Chr * Localization | Penetrance |
|---|---|---|---|
| CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A | 9p21.3 | High |
| CDK4 | Cyclin-Dependent Kinase 4 | 12q14.1 | High |
| BAP1 | BRCA1-Associated Protein 1 | 3p21.1 | High |
| TERT | Telomerase Reverse Transcriptase | 5p15.33 | High |
| POT1 | Protection of Telomeres 1 | 7q31.33 | High |
| ACD | ACD Shelterin Complex Subunit and Telomerase Recruitment Factor | 16q22.1 | High |
| TERF2IP | TERF2-Interacting Protein | 16q23.1 | High |
| MC1R | Melanocortin 1 Receptor | 16q24.3 | Moderate |
| MITF | Melanocyte-Inducing Transcription Factor | 3p13 | Moderate |
| MGMT | O-6-Methylguanine-DNA Methyltransferase | 10q26.3 | Low |
| TPCN2 | Two Pore Segment Channel 2 | 11q13.3 | Low |
| ASIP | Agouti Signaling Protein | 20q11.22 | Low |
| KITLG | KIT Ligand | 12q21.32 | Low |
| SLC24A5 | Solute Carrier Family 24 Member 5 | 15q21.1 | Low |
| SLC45A2 | Solute Carrier Family 45 Member 2 | 5p13.2 | Low |
| TYR | Tyrosinase | 11q14.3 | Low |
| IRF4 | Interferon Regulatory Factor 4 | 6p25.3 | Low |
| OCA2 | OCA2 Melanosomal Transmembrane Protein | 15q12-q13.1 | Low |
| TYRP1 | Tyrosinase-Related Protein 1 | 9p23 | Low |
| CYP1B1 | Cytochrome P450 family 1 subfamily B member 1 | 2p22.2 | Low |
| ARNT | Aryl Hydrocarbon Receptor Nuclear Translocator | 1q21.3 | Low |
| CDKAL1 | CDKAL1 threonylcarbamoyladenosine tRNA methylthiotransferase | 6p22.3 | Low |
| AGR3 | Anterior Gradient 3, protein disulfide isomerase family member | 7p21.1 | Low |
| TMEM38B | Transmembrane protein 38B | 9q31.2 | Low |
| ZNF462 | Zinc Finger protein 462 | 9q31.2 | Low |
| RAD23B | RAD23 nucleotide excision repair protein B | 9q31.2 | Low |
| OBFC1 (STN1) | STN1 subunit of CST complex | 10q24.33 | Low |
| SH3PXD2A | SH3 and PX domains 2A | 10q24.33 | Low |
| CCND1 | Cyclin D1 | 11q13.3 | Low |
| HERC2 | HECT and RLD domain containing E3 ubiquitin protein ligase 2 | 15q13.1 | Low |
| GOLM1 | Golgi membrane protein 1 | 9q21.33 | Low |
| TP53AIP1 | Tumor Protein p53 regulated Apoptosis Inducing Protein 1 | 11q24.3 | Low |
| IL-10 | Interleukin 10 | 1q32.1 | Low |
| IL-1β | Interleukin 1 Beta | 2q14.1 | Low |
| TNF-α | Tumor Necrosis Factor | 6p21.33 | Low |
| Gene | Cancer Risk Management |
|---|---|
| CDKN2A |
|
| CDK4 |
|
| BAP1 |
|
| POT1 |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarano, C.; Veneruso, I.; D’Argenio, V. Genetic Landscape of Familial Melanoma. Genes 2025, 16, 857. https://doi.org/10.3390/genes16080857
Scarano C, Veneruso I, D’Argenio V. Genetic Landscape of Familial Melanoma. Genes. 2025; 16(8):857. https://doi.org/10.3390/genes16080857
Chicago/Turabian StyleScarano, Carmela, Iolanda Veneruso, and Valeria D’Argenio. 2025. "Genetic Landscape of Familial Melanoma" Genes 16, no. 8: 857. https://doi.org/10.3390/genes16080857
APA StyleScarano, C., Veneruso, I., & D’Argenio, V. (2025). Genetic Landscape of Familial Melanoma. Genes, 16(8), 857. https://doi.org/10.3390/genes16080857