
Expanding the Phenotypic Spectrum Associated with DPH5-Related Diphthamide Deficiency

 , , , , ,
, , , , ,  , , , , and
, , , , and
Abstract
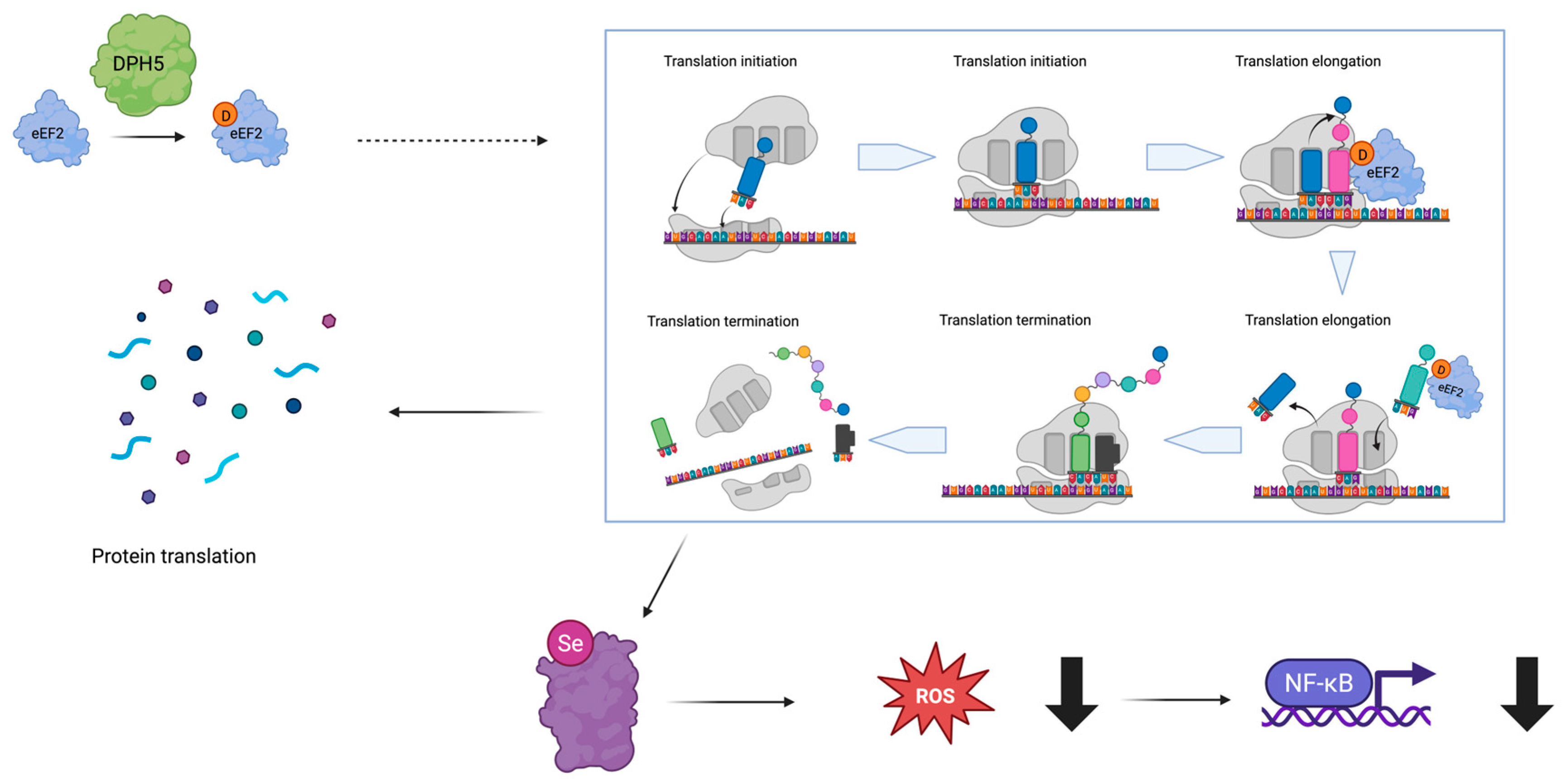
1. Introduction
2. Materials and Methods
3. Results
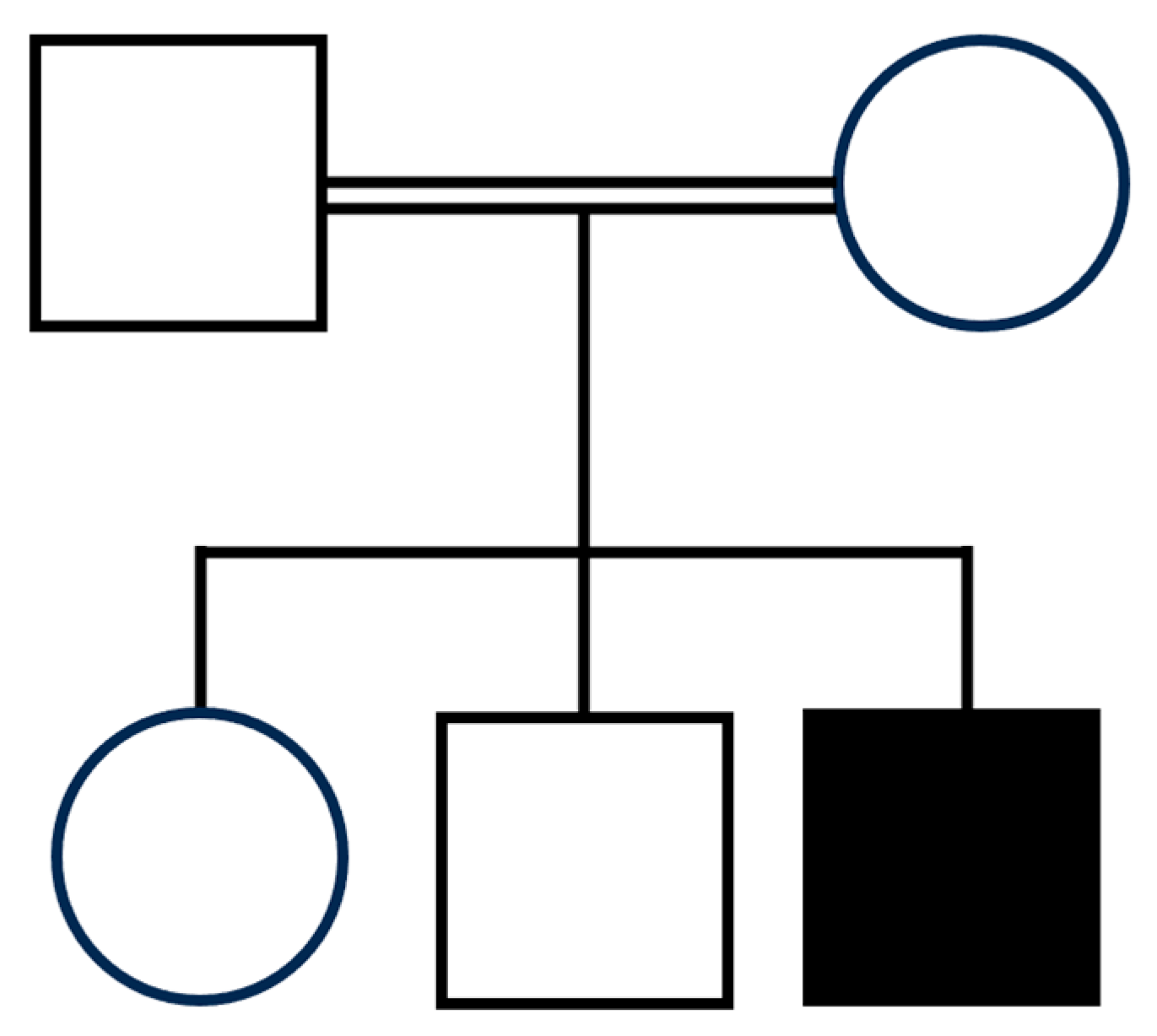
3.1. Clinical Assessment
3.2. Computational Facial Analysis
3.3. Molecular Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morris-Rosendahl, D.J.; Crocq, M.-A. Neurodevelopmental disorders—The history and future of a diagnostic concept. Dialogues Clin. Neurosci. 2020, 22, 65–72. [Google Scholar] [CrossRef]
- AlMutiri, R.; Malta, M.; Shevell, M.I.; Srour, M. Evaluation of Individuals with Non-Syndromic Global Developmental Delay and Intellectual Disability. Children 2023, 10, 414. [Google Scholar] [CrossRef]
- Fernandez, B.A.; Scherer, S.W. Syndromic autism spectrum disorders: Moving from a clinically defined to a molecularly defined approach. Dialogues Clin. Neurosci. 2017, 19, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Brunet, T.; Jech, R.; Brugger, M.; Kovacs, R.; Alhaddad, B.; Leszinski, G.; Riedhammer, K.M.; Westphal, D.S.; Mahle, I.; Mayerhanser, K.; et al. De novo variants in neurodevelopmental disorders-experiences from a tertiary care center. Clin. Genet. 2021, 100, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Kampen, K.R.; Sulima, S.O.; Vereecke, S.; De Keersmaecker, K. Hallmarks of ribosomopathies. Nucleic Acids Res. 2020, 48, 1013–1028. [Google Scholar] [CrossRef]
- Nakhoul, H.; Ke, J.; Zhou, X.; Liao, W.; Zeng, S.X.; Lu, H. Ribosomopathies: Mechanisms of Disease. Clin. Med. Insights Blood Disord. 2014, 7, 7–14. [Google Scholar] [CrossRef]
- Ütkür, K.; Mayer, K.; Khan, M.; Manivannan, T.; Schaffrath, R.; Brinkmann, U. DPH1 and DPH2 variants that confer susceptibility to diphthamide deficiency syndrome in human cells and yeast models. Dis. Model. Mech. 2023, 16, dmm050207. [Google Scholar] [CrossRef]
- Su, X.; Lin, Z.; Lin, H. The biosynthesis and biological function of diphthamide. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 515–521. [Google Scholar] [CrossRef]
- Hörberg, J.; Saenz-Mendez, P.; Eriksson, L.A. QM/MM Studies of Dph5—A Promiscuous Methyltransferase in the Eukaryotic Biosynthetic Pathway of Diphthamide. J. Chem. Inf. Model. 2018, 58, 1406–1414. [Google Scholar] [CrossRef]
- Schaffrath, R.; Abdel-Fattah, W.; Klassen, R.; Stark, M.J.R. The diphthamide modification pathway from Saccharomyces cerevisiae—revisited. Mol. Microbiol. 2014, 94, 1213–1226. [Google Scholar] [CrossRef]
- Loucks, C.M.; Parboosingh, J.S.; Shaheen, R.; Bernier, F.P.; McLeod, D.R.; Seidahmed, M.Z.; Puffenberger, E.G.; Ober, C.; Hegele, R.A.; Boycott, K.M.; et al. Matching Two Independent Cohorts Validates DPH1 as a Gene Responsible for Autosomal Recessive Intellectual Disability with Short Stature, Craniofacial, and Ectodermal Anomalies. Hum. Mutat. 2015, 36, 1015–1019. [Google Scholar] [CrossRef]
- Makrythanasis, P.; Guipponi, M.; Santoni, F.A.; Zaki, M.; Issa, M.Y.; Ansar, M.; Hamamy, H.; Antonarakis, S.E. Exome sequencing discloses KALRN homozygous variant as likely cause of intellectual disability and short stature in a consanguineous pedigree. Hum. Genom. 2016, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Hawer, H.; Mendelsohn, B.A.; Mayer, K.; Kung, A.; Malhotra, A.; Tuupanen, S.; Schleit, J.; Brinkmann, U.; Schaffrath, R. Diphthamide-deficiency syndrome: A novel human developmental disorder and ribosomopathy. Eur. J. Hum. Genet. 2020, 28, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.P.; Grimsrud, K.; Lanoue, L.; Egense, A.; Willis, B.; Hörberg, J.; AlAbdi, L.; Mayer, K.; Ütkür, K.; Monaghan, K.G.; et al. A novel DPH5-related diphthamide-deficiency syndrome causing embryonic lethality or profound neurodevelopmental disorder. Genet. Med. 2022, 24, 1567–1582. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1098360022007043 (accessed on 1 June 2024). [CrossRef]
- Mayer, K.; Mundigl, O.; Kettenberger, H.; Birzele, F.; Stahl, S.; Pastan, I.; Brinkmann, U. Diphthamide affects selenoprotein expression: Diphthamide deficiency reduces selenocysteine incorporation, decreases selenite sensitivity and pre-disposes to oxidative stress. Redox Biol. 2019, 20, 146–156. [Google Scholar] [CrossRef]
- Kaltschmidt, C.; Greiner, J.F.W.; Kaltschmidt, B. The Transcription Factor NF-κB in Stem Cells and Development. Cells 2021, 10, 2042. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.-C.; Bar-Haim, A.; Moosa, S.; Ehmke, N.; Gripp, K.W.; Pantel, J.T.; Danyel, M.; Mensah, M.A.; Horn, D.; Rosnev, S.; et al. GestaltMatcher facilitates rare disease matching using facial phenotype descriptors. Nat. Genet. 2022, 54, 349–357. [Google Scholar] [CrossRef]
- Motta, M.; Pannone, L.; Pantaleoni, F.; Bocchinfuso, G.; Radio, F.C.; Cecchetti, S.; Ciolfi, A.; Di Rocco, M.; Elting, M.W.; Brilstra, E.H.; et al. Enhanced MAPK1 Function Causes a Neurodevelopmental Disorder within the RASopathy Clinical Spectrum. Am. J. Hum. Genet. 2020, 107, 499–513. [Google Scholar] [CrossRef]
- Radio, F.C.; Pang, K.; Ciolfi, A.; Levy, M.A.; Hernández-García, A.; Pedace, L.; Pantaleoni, F.; Liu, Z.; de Boer, E.; Jackson, A.; et al. SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am. J. Hum. Genet. 2021, 108, 502–516. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 1110. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef]
- Popov, I.K.; Hiatt, S.M.; Whalen, S.; Keren, B.; Ruivenkamp, C.; van Haeringen, A.; Chen, M.J.; Cooper, G.M.; Korf, B.R.; Chang, C. A YWHAZ Variant Associated with Cardiofaciocutaneous Syndrome Activates the RAF-ERK Pathway. Front. Physiol. 2019, 10, 388. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Brajanovski, N.; Chan, K.T.; Xuan, J.; Pearson, R.B.; Sanij, E. Ribosomal proteins and human diseases: Molecular mechanisms and targeted therapy. Signal Transduct. Target. Ther. 2021, 6, 323. [Google Scholar] [CrossRef]
- Tidyman, W.E.; Rauen, K.A. The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 2009, 19, 230–236. [Google Scholar] [CrossRef]
- Tartaglia, M.; Gelb, B.D. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: Phenotypic spectrum and molecular mechanisms. Ann. N. Y. Acad. Sci. 2010, 1214, 99–121. [Google Scholar] [CrossRef]
- Tartaglia, M.; Aoki, Y.; Gelb, B.D. The molecular genetics of RASopathies: An update on novel disease genes and new disorders. Am. J. Med. Genet. C Semin. Med. Genet. 2022, 190, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Tsuda-Sakurai, K.; Kimura, M.; Miura, M. Diphthamide modification of eEF2 is required for gut tumor-like hyperplasia induced by oncogenic Ras. Genes. Cells 2020, 25, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Al Hmada, Y.; Brodell, R.T.; Kharouf, N.; Flanagan, T.W.; Alamodi, A.A.; Hassan, S.Y.; Shalaby, H.; Hassan, S.L.; Haikel, Y.; Megahed, M. Mechanisms of Melanoma Progression and Treatment Resistance: Role of Cancer Stem-like Cells. Cancers 2024, 16, 470. [Google Scholar] [CrossRef] [PubMed]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol. Cancer Ther. 2017, 16, 2351–2363. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Features | DPH5 Variant and Inheritance Model | Age | Sex | Positive Family History | Weight (SD If p < 3rd p) | Height (SD If p < 3rd p) | Head Circumference (SD if p < 3rd p) | Facial Features | Prenatal/Birth History | Perinatal History | Global Developmental Delay | Independent Ambulation | Language | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Present case | Hz c.779A>G (p.His260Arg) | 10 y | M | No | 3rd p (−2 SD) | <3rd p (−3.7 SD) | 50th–75th p | Coarse facial features, bitemporal narrowing, bilateral frontal and parietal bossing, high forehead, triangular face, synophrys, epicanthus, hypertelorism, bilateral ptosis, blue sclerae, depressed nasal bridge, low-set ears, overfolded dysmorphic ears with thick ear lobe, long deep philtrum, thick lips, pointed chin. | Normal pregnancy, Cesarean section for previous cesarean section. | Feeding difficulties | Yes | Yes, milestone reached at 6 years of age | Non verbal | |
| Patient 1 [15] | Hz c.779A>G (p.His260Arg) | 10 y | F | One previous live birth with mortality at 1 week of age. Similarly affected sibling (patient 2). | Failure to thrive, 10th p with G tube feeds. | <3rd p (−2.5 SD) | 14th p | Broad forehead, bitemporal narrowing, sparse eyebrows, epicanthal folds, deep-set eyes, broad nasal bridge, rounded tip of nose, downturned corners of mouth, high arched palate, triangular chin, dental caries. | Decreased fetal movement in pregnancy, born at 42 weeks. | Neonatal intensive care unit stay for hypoxia | Yes | Yes, but limited | Non verbal | |
| Patient 2 [15] | Hz c.779A>G (p.His260Arg) | 8 y | M | One previous live birth with mortality at age 1 week. Similarly affected sibling (patient 1). | 70th p | 10th–25th p | 23rd p | Broad forehead, bitemporal narrowing, sparse eyebrows, epicanthal folds, wide palpebral fissures, broad nasal bridge, rounded nasal tip, downturned corners of mouth, triangular chin, multiple dental caries. | Normal pregnancy, normal spontaneous vaginal delivery. | No issues | Yes | No | Non verbal | |
| Patient 3 [15] | Compound het c.619C>T (p.Arg207 *) and c.329A>G (p.Asn110Ser) | 9 y | M | Similarly affected sibling (patient 4). | <3rd p (−5SD) | <3rd p (−4.99 SD) | 13th p | High forehead, high anterior hairline, depressed midface, upslanting eyes, sparse eyelashes, mild epicanthal folds, thick alveolar ridges. | Decreased fetal movement, excess fluid at delivery. | Hospital stay for 3 weeks for poor feeding | Yes | No | Non verbal | |
| Patient 4 [15] | Compound het c.619C>T (p.Arg207 *) and c.329A>G (p.Asn110Ser) | 1 y | F | Similarly affected sibling (patient 3). | <3rd p (−3.45 SD) | <3rd p (−3.06 SD) | 32nd p | Prominent forehead, depressed midface, broad alveolar ridges, faint eyebrows, upslanting eyes. | Excess fluid at delivery. | No issues | Yes | NA for age | Non verbal | |
| Patient 5 [15] | Hz c.521dupA (p.Asn174LysfTer10) | 11 d | F | Spontaneous miscarriages, one antepartum stillbirth, and one neonatal death. Microphthalmia, hypertelorism, IUGR, TOF, hydrocephalus, and cerebellar hypoplasia in dead babies. | BW 800 gm | NA | NA | Broad forehead, bitemporal narrowing, sparse eyebrows, epicanthal folds, wide palpebral fissures, deep-set eyes, broad nasal bridge, rounded nasal tip, downturned corners of mouth, triangular chin, micrognathia. | Polyhydramnios. Preterm baby delivered via Cesarean section for breech presentation | Cyanosis | NA | NA for age | NA | |
| Features | Intellectual disability | Behavioral concerns | Nervous system dysfunction | Brain imaging studies (age) | EEG | Hearing | Cardiac system | Respiratory system | Gastrointestinal system | Endocrinology | Musculoskeletal | Urogenital | Ophthalmology | Dermatology |
| Present case | Profound | Happy disposition | Seizures, Diffuse hypotonia, sleep difficulties | Brain MRI (3 y): corpus callosum hypoplasia, inferior vermis hypoplasia, enlarged cisterna magna. | Poor organization with multifocal spike and wave discharges, prevalent in the temporo-posterior regions. | Slightly altered BAEP | Normal | Recurrent episodes of cyanosis and apnoea. Frequent infections and moderate obstructive sleep apnea. | GERD | Short stature (started GH replacement therapy) | Kyphosis, joint laxity, hypotonia, and global hypotrophy, pes planus. | Incontinence | Strabismus, abnormal ocular movements (nystagmus) | Normal |
| Patient 1 [15] | Profound | Screaming episodes | Congenital hypotonia, cerebral palsy, sleep difficulties | Brain MRI (1 y): brain atrophy. CT scan (8 y): within normal limits. | Abnormal, continuous slowing consistent with mild to moderate encephalopathy. | Normal BAEP | Trivial mitral prolapse and regurgitation, normal ECG | Normal | Feeding difficulties, dysphagia, GJ-tube fed, bowel incontinence | Short stature | Lean muscle mass, small hands and feet, tapered fingers, brachydactyly of the third toe, bilateral. | Urinary incontinence | Fixes and follows | Normal |
| Patient 2 [15] | Profound | Happy disposition | Seizures, spastic quadriplegia, cerebral palsy, contractures, weakness, DTRs 3+, plantars upgoing | Brain MRI (5 y): within normal limits. | Abnormal, consistent with generalized disturbance of cerebral function with multifocal epileptogenic brain abnormality. | Normal BAEP | Trivial mitral prolapse and regurgitation, sinus tachycardia at ECG | Dyspnea, obstructive sleep apnea, asthma, recurrent aspiration, pneumonia, and oxygen therapy at night. | GJ-tube dependent, hematemesis | Short stature | Lean muscle mass, tapered fingers, pes planus, brachydactyly of 4 toes other than the big toe, bilateral. | Urinary incontinence | Ocular melanocytosis, pathologic high myopia-11 DS, fixes and follows | Normal |
| Patient 3 [15] | Profound | NA | Seizure, appendicular spasticity, central hypotonia, motor regression | Brain MRI (NA): Focal lesion in white matter of left inferior frontal gyrus, vertically oriented hippocampi, prominence of ventricular system. | Abnormal. | Normal, BAEP NA | Congenital aortic dilatation | Breath-holding spells. | G tube | Short stature | Rocker bottom feet, hyperextensible joints, tapered fingers, right single crease, small hands, and feet. | No known issues | Gray sclera with ocular melanocytosis, strabismus requiring surgery | Extensible skin |
| Patient 4 [15] | NA for age | NA | Myoclonic seizures, central hypotonia, and appendicular hypertonia | Brain MRI (NA): Diffuse paucity of white matter, cerebellar vermian hypoplasia. | Abnormal, consistent with epileptic myoclonus. | Normal, BAEP NA | Aortic dilatation | Normal | GERD | Short stature | Tapered fingers. | No known issues | Gray sclera, cortical visual impairment | Extensible skin, pale |
| Patient 5 [15] | NA for age | NA | Congenital hypotonia | Prenatal ultrasound: “Strawberry head”. Brain MRI (NA): bilateral minimal tentorial subdural hemorrhage and enlarged cisterna magna. | NA | NA | Multiple muscular VSD and ASD, hypoplastic pulmonary artery, pericardial effusion | Normal | Bowel perforation at 10 days of life, died the following day. | NA | NA | NA | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Politano, D.; Mancini, C.; Celario, M.; Radio, F.C.; D'Abrusco, F.; Garau, J.; Kalantari, S.; Visani, G.; Carbonera, S.; Gana, S.; et al. Expanding the Phenotypic Spectrum Associated with DPH5-Related Diphthamide Deficiency. Genes 2025, 16, 799. https://doi.org/10.3390/genes16070799
Politano D, Mancini C, Celario M, Radio FC, D'Abrusco F, Garau J, Kalantari S, Visani G, Carbonera S, Gana S, et al. Expanding the Phenotypic Spectrum Associated with DPH5-Related Diphthamide Deficiency. Genes. 2025; 16(7):799. https://doi.org/10.3390/genes16070799
Chicago/Turabian StylePolitano, Davide, Cecilia Mancini, Massimiliano Celario, Francesca Clementina Radio, Fulvio D'Abrusco, Jessica Garau, Silvia Kalantari, Gaia Visani, Simone Carbonera, Simone Gana, and et al. 2025. "Expanding the Phenotypic Spectrum Associated with DPH5-Related Diphthamide Deficiency" Genes 16, no. 7: 799. https://doi.org/10.3390/genes16070799
APA StylePolitano, D., Mancini, C., Celario, M., Radio, F. C., D'Abrusco, F., Garau, J., Kalantari, S., Visani, G., Carbonera, S., Gana, S., Ferilli, M., Chiriatti, L., Cappelletti, C., Ellena, K., Prodi, E., Borgatti, R., Valente, E. M., Orcesi, S., Tartaglia, M., & Sirchia, F. (2025). Expanding the Phenotypic Spectrum Associated with DPH5-Related Diphthamide Deficiency. Genes, 16(7), 799. https://doi.org/10.3390/genes16070799