
Somatic Mutations Associated with Aldosterone-Producing Adenomas (APAs)


Abstract
1. Introduction
2. Literature Search Strategy
3. Background
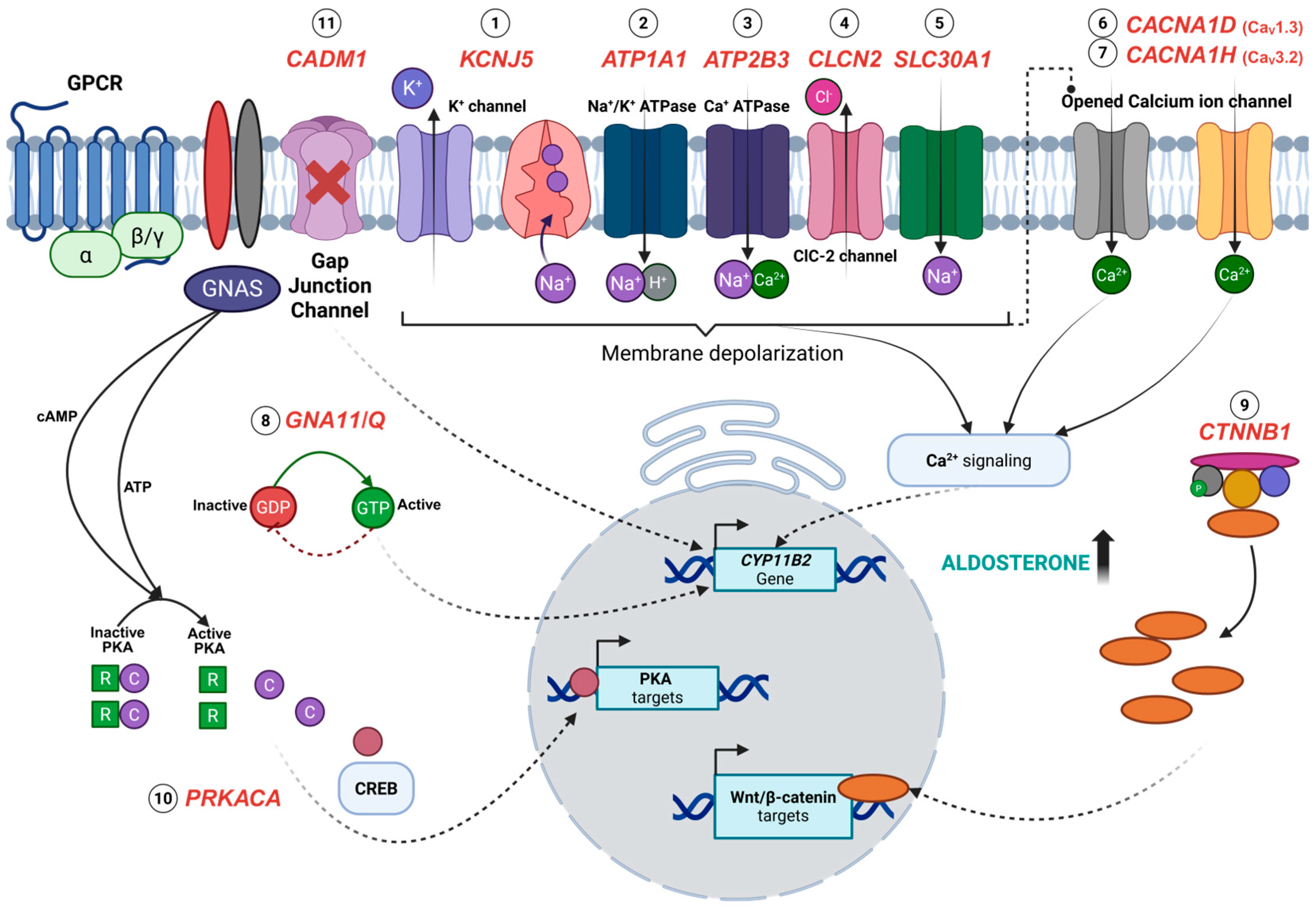
4. Somatic Mutations Associated with APAs
4.1. KCNJ5
4.2. CACNA1D
4.3. ATP1A1 and ATP2B3
4.4. CTNNB1
4.5. GNA11 and GNAQ
4.6. CACNA1H
4.7. CLCN2
4.8. CADM1
4.9. SLC30A1
4.10. PRKACA
5. Ethnic and Gender Variations in APA Mutations

{kind=link}
| Gene | Male | Female | Sample Size, n | References |
|---|---|---|---|---|
| KCNJ5 mutations | 357 (30%) | 656 (56%) | 1180 | |
| p.Gly151Arg; p.Leu168Arg | 1 (1%) | 7 (88%) | 8 | [11] |
| p.Gly151Arg; p.Leu168Arg | 31 (24%) | 97 (74%) | 128 | [12] |
| p.Glu145Gln; p.Gly151Arg; p.Leu168Arg | 24 (8%) | 112 (39%) | 287 | [22] |
| p.Gly151Arg; p.Leu168Arg; p.Ile157del | 1 (12.5%) | 3 (37.5%) | 8 | [23] |
| p.Gly151Arg; p.Leu168Arg | 7 (47%) | 8 (53%) | 15 | [25] |
| p.Gly151Arg; p.Leu168Arg | 5 (50%) | 5 (50%) | 10 | [26] |
| p.Trp126Arg; p.Gly151Arg; p.Thr158Ala; p.Leu168Arg | 21 (28%) | 53 (72%) | 74 | [28] |
| p.Arg115Trp; p.Glu145Gln; p.Gly151Arg; p.Leu168Arg; p.Glu246Gly | 9 (35%) | 17 (65%) | 26 | [29] |
| p.Glu145Gln; p.Gly151Arg; p.Ile157del; p.Thr158Ala; p.Leu168Arg | 25 (33%) | 50 (67%) | 75 | [30] |
| p.Gly151Arg; p.Ile157del; p.Thr158Ala; p.Leu168Arg | 62 (68%) | 26 (28.6%) | 91 | [31] |
| p.Thr148_Thr149insArg; p.Gly151Arg; p.Thr158Ala; p.Leu168Arg | 56 (43%) | 73 (57%) | 129 | [32] |
| NA | 16 (17%) | 76 (83%) | 92 | [33] |
| p.Glu145Lys; p.Gly151Arg; p.Leu168Arg | 5 (18%) | 5 (18%) | 28 | [34] |
| p.Gly151Arg; p.Ile157del; p.Thr158Ala; p.Leu168Arg | 48 (41.4%) | 68 (58.6%) | 116 | [35] |
| p.[Thr148Ile;Thr149Ser]; p.Thr149delinsThrThr; p.Thr149delinsMetAla; p.Gly151Arg; p.Leu168Arg | 5 (13%) | 20 (57%) | 25 | [36] |
| p.Glu145Gln; p.Gly151Arg; p.Thr158Ala; p.Leu168Arg | 41 (60%) | 36 (95%) | 68 | [37] |
| CACNA1D mutations | 63 (19%) | 25 (8%) | 331 | |
| pVal401Leu; p.Gly403Arg; p.Phe747Leu; p.Val1353Met | 3 (60%) | 2 (40%) | 5 | [15] |
| p.Gly403Arg; p.Ile770Met | 3 (7%) | 2 (5%) | 43 | [27] |
| p.Val259Asp; p.Gly403Arg; p.Ser652Leu; p.Leu655Pro; p.Tyr741Cys; p.Phe747Val; p.Phe747Leu; p.Ile750Met; p.Ile750Phe; p.Val979Asp; p.Val981Asn; p.Ala998Ile; p.Ala998Val; p.Val1151Phe; p.Ile1152Asn; p.Pro1336Arg; p.Val1338Met; p.Met1354Ile | 18 (67%) | 9 (33%) | 27 | [28] |
| p.Gly403Arg | 0 | 1 (0.8%) | 129 | [32] |
| p.Gly403Arg; p.Phe747Leu; p.Arg990His; p.Val1153Gly | 4 (14%) | 0 | 28 | [34] |
| p.Val309Ala; p.Val401Leu; p.Gly403Arg; p.Arg619Pro; p.Ser652Leu; p.Phe747Val/Leu/Cys; p.Ile750Phe/Met; p.Arg990Gly; p.Arg993Thr; p.Ala998Val; p.Cys1007Arg; p.Ile1015Ser; p.Val1151Phe | 21 (55%) | 10 (29%) | 31 | [36] |
| p.Gly403Arg; p.Ser652Leu; p.Phe747Val; p.Ser969Leu; p.Arg990His; p.Ala998Val/Ile; p.Ile1015Thr; p.Val1338Met | 14 (21%) | 1 (3%) | 68 | [37] |
| ATP1A1 mutations | 38 (11%) | 5 (2%) | 341 | |
| p.Met102_Leu103del; p.Met102_Ile106del; p.Leu103_Leu104del; p.Leu104Arg; p.Phe956_Glu961del; p.Phe959_Glu961del; p.Glu960_Leu964del | 8 (80%) | 2 (20%) | 10 | [15] |
| p.Gly99Arg; p.Phe100_Leu104del; p.Leu104Arg; p.Val332Gly | 8 (89%) | 1 (11%) | 9 | [28] |
| p.Leu104Arg | 2 (2.2%) | 0 | 91 | [31] |
| p.Met102_Leu103del; p.Leu104Arg | 4 (3%) | 0 | 129 | [32] |
| p.Phe100_Leu104del; p.Leu104Arg | 6 (21%) | 1 (4%) | 28 | [34] |
| p.Leu104Arg; p.Ile955_Glu960delinsLys | 5 (13%) | 1 (3%) | 6 | [36] |
| p.Leu104Arg; p.Phe959_Glu961delinsLeu; p.Glu960_Ala965delinsAlaLeuVal | 5 (7%) | 0 | 68 | [37] |
| ATP2B3 mutations | 10 (3%) | 7 (2%) | 299 | |
| p.Thr423_Leu425del; p.Val424_Leu425del; p.Val424_Val426del; p.Val426_Val429del | 2 (40%) | 3 (60%) | 5 | [15] |
| p.Leu424_Val425del; p.Leu425_Val426del; p.Val426_Val427del | 1 (33%) | 2 (67%) | 3 | [28] |
| p.Tyr410Asp | 0 | 1 (1%) | 91 | [31] |
| p.Val422-Val426delinsSerThrLeu | 1 (1%) | 0 | 129 | [32] |
| p.Val424_Leu425del | 2 (5%) | 1 (3%) | 3 | [36] |
| p.Val424_Leu425del; p.Leu425_Val426del | 4 (6%) | 0 | 68 | [37] |
| CTNNB1 mutations | 6 (33%) | 12 (67%) | 18 | |
| p.Thr41Ala; p.Ser45Phe; p.Ser45Pro | 4 (40%) | 6 (60%) | 10 | [33] |
| p.Ser45Phe; p.Ser45Pro | 2 (25%) | 6 (75%) | 8 | [35] |
| GNA11 mutations (co-occurring with CTNNB1) | 1 (4%) | 10 (37%) | 27 | |
| p.Gln209Pro with p.Gly34Arg/p.Ser45Phe/p.Ser45Pro; p.Gln209His with p.Ser33Cys/p.Thr41Ala/p.Ser45Phe | 1 (4%) | 10 (37%) | 27 | [46] |
| GNAQ mutations (co-occurring with CTNNB1) | 0 | 5 (18%) | 27 | |
| p.Gln209His with p.Gly34Glu; p.Gln209Leu with p.Gly34Arg | 0 | 5 (18%) | 27 | [46] |
| CACNA1H mutations | 2 (3%) | 1 (1%) | 75 | |
| p.Ile1430Thr | 2 (3%) | 1 (1%) | 75 | [38] |
| CLCN2 mutations | 2 (1%) | 2 (1%) | 207 | |
| p.Gly24Asp | 0 | 1 (8.3%) | 12 | [39] |
| p.Gly24Asp | 1 (1.3%) | 0 | 80 | [40] |
| p.Gly24Asp; c.64-2_74del | 1 (0.9%) | 1 (0.9%) | 115 | [41] |
| CADM1 mutations | 4 (10%) | 2 (5%) | 40 | |
| p.Gly379Asp; p.Val380Asp | 4 (10%) | 2 (5%) | 40 | [43] |
| SLC30A1 mutations | 5 (3%) | 0 | 186 | |
| p.Leu49_Leu55del; p.Leu51_Ala57del | 5 (3%) | 0 | 186 | [42] |
| PRKACA mutations | 0 | 3 (2.1%) | 142 | |
| p.His88Asp; p.Leu206Arg | 0 | 2 (2%) | 122 | [44] |
| p.Leu206Arg | 0 | 1 (5%) | 20 | [45] |
6. Clinical Implications of the Somatic Mutations
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization (WHO). Hypertension 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/hypertension (accessed on 6 December 2024).
- Tomaschitz, A.; Ritz, E.; Pieske, B.; Rus-Machan, J.; Kienreich, K.; Verheyen, N.; Gaksch, M.; Grübler, M.; Fahrleitner-Pammer, A.; Mrak, P.; et al. Aldosterone and parathyroid hormone interactions as mediators of metabolic and cardiovascular disease. Metab. Clin. Exp. 2014, 63, 20–31. [Google Scholar] [CrossRef]
- Milliez, P.; Girerd, X.; Plouin, P.F.; Blacher, J.; Safar, M.E.; Mourad, J.J. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J. Am. Coll. Cardiol. 2005, 45, 1243–1248. [Google Scholar] [CrossRef]
- Hegde, S.; Ahmed, I.; Aeddula, N. Secondary Hypertension. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ebbehoj, A.; Li, D.; Kaur, R.J.; Zhang, C.; Singh, S.; Li, T.; Atkinson, E.; Achenbach, S.; Khosla, S.; Arlt, W.; et al. Epidemiology of adrenal tumours in Olmsted County, Minnesota, USA: A population-based cohort study. Lancet Diabetes Endocrinol. 2020, 8, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Reimondo, G.; Castellano, E.; Grosso, M.; Priotto, R.; Puglisi, S.; Pia, A.; Pellegrino, M.; Borretta, G.; Terzolo, M. Adrenal Incidentalomas are Tied to Increased Risk of Diabetes: Findings from a Prospective Study. J. Clin. Endocrinol. Metab. 2020, 105, dgz284. [Google Scholar] [CrossRef]
- Yizhak, K.; Aguet, F.; Kim, J.; Hess, J.M.; Kübler, K.; Grimsby, J.; Frazer, R.; Zhang, H.; Haradhvala, N.J.; Rosebrock, D.; et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019, 364, eaaw0726. [Google Scholar] [CrossRef] [PubMed]
- Omata, K.; Anand, S.K.; Hovelson, D.H.; Liu, C.J.; Yamazaki, Y.; Nakamura, Y.; Ito, S.; Satoh, F.; Sasano, H.; Rainey, W.E.; et al. Aldosterone-Producing Cell Clusters Frequently Harbor Somatic Mutations and Accumulate With Age in Normal Adrenals. J. Endocr. Soc. 2017, 1, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Azizan, E.A.; Poulsen, H.; Tuluc, P.; Zhou, J.; Clausen, M.V.; Lieb, A.; Maniero, C.; Garg, S.; Bochukova, E.G.; Zhao, W.; et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat. Genet. 2013, 45, 1055–1060. [Google Scholar] [CrossRef]
- Beuschlein, F.; Boulkroun, S.; Osswald, A.; Wieland, T.; Nielsen, H.N.; Lichtenauer, U.D.; Penton, D.; Schack, V.R.; Amar, L.; Fischer, E.; et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat. Genet. 2013, 45, 440–444. [Google Scholar] [CrossRef]
- Choi, M.; Scholl, U.I.; Yue, P.; Björklund, P.; Zhao, B.; Nelson-Williams, C.; Ji, W.; Cho, Y.; Patel, A.; Men, C.J.; et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011, 331, 768–772. [Google Scholar] [CrossRef]
- Boulkroun, S.; Beuschlein, F.; Rossi, G.P.; Golib-Dzib, J.F.; Fischer, E.; Amar, L.; Mulatero, P.; Samson-Couterie, B.; Hahner, S.; Quinkler, M.; et al. Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension 2012, 59, 592–598. [Google Scholar] [CrossRef]
- Conn, J.W. Primary aldosteronism. J. Lab. Clin. Med. 1955, 45, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Amar, L.; Plouin, P.F.; Steichen, O. Aldosterone-producing adenoma and other surgically correctable forms of primary aldosteronism. Orphanet J. Rare Dis. 2010, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Åkerström, T.; Willenberg, H.S.; Cupisti, K.; Ip, J.; Backman, S.; Moser, A.; Maharjan, R.; Robinson, B.; Iwen, K.A.; Dralle, H.; et al. Novel somatic mutations and distinct molecular signature in aldosterone-producing adenomas. Endocr.-Relat. Cancer. 2015, 22, 735–744. [Google Scholar] [CrossRef]
- Fogari, R.; Preti, P.; Zoppi, A.; Rinaldi, A.; Fogari, E.; Mugellini, A. Prevalence of primary aldosteronism among unselected hypertensive patients: A prospective study based on the use of an aldosterone/renin ratio above 25 as a screening test. Hypertens. Res. 2007, 30, 111–117. [Google Scholar] [CrossRef]
- Rossi, G.P.; Bernini, G.; Caliumi, C.; Desideri, G.; Fabris, B.; Ferri, C.; Ganzaroli, C.; Giacchetti, G.; Letizia, C.; Maccario, M.; et al. A prospective study of the prevalence of primary aldosteronism in 1125 hypertensive patients. J. Am. Coll. Cardiol. 2006, 48, 2293–2300. [Google Scholar] [CrossRef]
- Savard, S.; Amar, L.; Plouin, P.F.; Steichen, O. Cardiovascular complications associated with primary aldosteronism: A controlled cross-sectional study. Hypertension 2013, 62, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Kang, V.J.; Pan, C.T.; Huang, J.Z.; Lin, Y.L.; Chang, Y.Y.; Tsai, C.; Chou, C.; Chen, Z.W.; Liao, C.; et al. KCNJ5 Somatic Mutation Is Associated With Higher Aortic Wall Thickness and Less Calcification in Patients With Aldosterone-Producing Adenoma. Front. Endocrinol. 2022, 13, 830130. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.W.; Lin, L.Y.; Hung, C.S.; Lin, Y.T.; Chang, Y.Y.; Wang, S.M.; Wu, V.C.; Wu, K.D.; Ho, Y.L.; Satoh, F.; et al. Time course and factors predicting arterial stiffness reversal in patients with aldosterone-producing adenoma after adrenalectomy: Prospective study of 102 patients. Sci. Rep. 2016, 6, 20862. [Google Scholar] [CrossRef]
- Lin, Y.H.; Lin, L.Y.; Chen, A.; Wu, X.M.; Lee, J.K.; Su, T.C.; Wu, V.C.; Chueh, S.C.; Lin, W.C.; Lo, M.T.; et al. Adrenalectomy improves increased carotid intima-media thickness and arterial stiffness in patients with aldosterone producing adenoma. Atherosclerosis 2012, 221, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Åkerström, T.; Crona, J.; Delgado Verdugo, A.; Starker, L.F.; Cupisti, K.; Willenberg, H.S.; Knoefel, W.T.; Saeger, W.; Feller, A.; Ip, J.; et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLoS ONE 2012, 7, e41926. [Google Scholar] [CrossRef]
- Azizan, E.A.; Lam, B.Y.; Newhouse, S.J.; Zhou, J.; Kuc, R.E.; Clarke, J.; Happerfield, L.; Marker, A.; Hoffman, G.J.; Brown, M.J. Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J. Clin. Endocrinol. Metab. 2012, 97, E819–E829. [Google Scholar] [CrossRef] [PubMed]
- Azizan, E.A.; Murthy, M.; Stowasser, M.; Gordon, R.; Kowalski, B.; Xu, S.; Brown, M.J.; O’Shaughnessy, K.M. Somatic mutations affecting the selectivity filter of KCNJ5 are frequent in 2 large unselected collections of adrenal aldosteronomas. Hypertension 2012, 59, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, R.; Yamada, M.; Nakajima, Y.; Satoh, T.; Hashimoto, K.; Shibusawa, N.; Ozawa, A.; Okada, S.; Rokutanda, N.; Takata, D.; et al. Expression and mutations of KCNJ5 mRNA in Japanese patients with aldosterone-producing adenomas. J. Clin. Endocrinol. Metab. 2012, 97, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, T.; Glomnes, N.; Strømsøy, S.; Knappskog, S.; Heie, A.; Akslen, L.A.; Grytaas, M.; Varhaug, J.E.; Gimm, O.; Brauckhoff, M. Outcome after surgery for primary hyperaldosteronism may depend on KCNJ5 tumor mutation status: A population-based study from Western Norway. Langenbeck’s Arch. Surg. 2013, 398, 869–874. [Google Scholar] [CrossRef]
- Scholl, U.I.; Goh, G.; Stölting, G.; de Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054. [Google Scholar] [CrossRef]
- Fernandes-Rosa, F.L.; Williams, T.A.; Riester, A.; Steichen, O.; Beuschlein, F.; Boulkroun, S.; Strom, T.M.; Monticone, S.; Amar, L.; Meatchi, T.; et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 2014, 64, 354–361. [Google Scholar] [CrossRef]
- Cheng, C.J.; Sung, C.C.; Wu, S.T.; Lin, Y.C.; Sytwu, H.K.; Huang, C.L.; Lin, S.H. Novel KCNJ5 mutations in sporadic aldosterone-producing adenoma reduce Kir3.4 membrane abundance. J. Clin. Endocrinol. Metab. 2015, 100, E155–E163. [Google Scholar] [CrossRef]
- Kitamoto, T.; Suematsu, S.; Matsuzawa, Y.; Saito, J.; Omura, M.; Nishikawa, T. Comparison of cardiovascular complications in patients with and without KCNJ5 gene mutations harboring aldosterone-producing adenomas. J. Atheroscler. Thromb. 2015, 22, 191–200. [Google Scholar] [CrossRef]
- Wu, V.C.; Huang, K.H.; Peng, K.Y.; Tsai, Y.C.; Wu, C.H.; Wang, S.M.; Yang, S.Y.; Lin, L.Y.; Chang, C.C.; Lin, Y.H.; et al. Prevalence and clinical correlates of somatic mutation in aldosterone producing adenoma-Taiwanese population. Sci. Rep. 2015, 5, 11396. [Google Scholar] [CrossRef]
- Zheng, F.F.; Zhu, L.M.; Nie, A.F.; Li, X.Y.; Lin, J.R.; Zhang, K.; Chen, J.; Zhou, W.L.; Shen, Z.J.; Zhu, Y.C.; et al. Clinical characteristics of somatic mutations in Chinese patients with aldosterone-producing adenoma. Hypertension 2015, 65, 622–628. [Google Scholar] [CrossRef]
- Åkerström, T.; Maharjan, R.; Willenberg, H.S.; Cupisti, K.; Ip, J.; Moser, A.; Stålberg, P.; Robinson, B.; Iwen, K.A.; Dralle, H.; et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci. Rep. 2016, 6, 19546. [Google Scholar] [CrossRef]
- Tan, G.C.; Negro, G.; Pinggera, A.; Tizen Laim, N.M.S.; Mohamed Rose, I.; Ceral, J.; Rsyka, A.; Chin, L.K.; Kamaruddin, N.A.; Mokhtar, N.M.; et al. Aldosterone-Producing Adenomas: Histopathology-Genotype Correlation and Identification of a Novel CACNA1D Mutation. Hypertension 2017, 70, 129–136. [Google Scholar] [CrossRef]
- Wu, V.C.; Wang, S.M.; Chueh, S.J.; Yang, S.Y.; Huang, K.H.; Lin, Y.H.; Wang, J.J.; Connolly, R.; Hu, Y.H.; Gomez-Sanchez, C.E.; et al. The prevalence of CTNNB1 mutations in primary aldosteronism and consequences for clinical outcomes. Sci. Rep. 2017, 7, 39121. [Google Scholar] [CrossRef]
- Nanba, K.; Omata, K.; Gomez-Sanchez, C.E.; Stratakis, C.A.; Demidowich, A.P.; Suzuki, M.; Thompson, L.D.R.; Cohen, D.L.; Luther, J.M.; Gellert, L.; et al. Genetic Characteristics of Aldosterone-Producing Adenomas in Blacks. Hypertension 2019, 73, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Nanba, K.; Yamazaki, Y.; Bick, N.; Onodera, K.; Tezuka, Y.; Omata, K.; Ono, Y.; Blinder, A.R.; Tomlins, S.A.; Rainey, W.E.; et al. Prevalence of Somatic Mutations in Aldosterone-Producing Adenomas in Japanese Patients. J. Clin. Endocrinol. Metab. 2020, 105, e4066–e4073. [Google Scholar] [CrossRef] [PubMed]
- Nanba, K.; Blinder, A.R.; Rege, J.; Hattangady, N.G.; Else, T.; Liu, C.J.; Tomlins, S.A.; Vats, P.; Kumar-Sinha, C.; Giordano, T.J.; et al. Somatic CACNA1H Mutation As a Cause of Aldosterone-Producing Adenoma. Hypertension 2020, 75, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Rosa, F.L.; Daniil, G.; Orozco, I.J.; Göppner, C.; El Zein, R.; Jain, V.; Boulkroun, S.; Jeunemaitre, X.; Amar, L.; Lefebvre, H.; et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat. Genet. 2018, 50, 355–361. [Google Scholar] [CrossRef]
- Dutta, R.K.; Arnesen, T.; Heie, A.; Walz, M.; Alesina, P.; Söderkvist, P.; Gimm, O. A somatic mutation in CLCN2 identified in a sporadic aldosterone-producing adenoma. Eur. J. Endocrinol. 2019, 181, K37–K41. [Google Scholar] [CrossRef]
- Rege, J.; Nanba, K.; Blinder, A.R.; Plaska, S.; Udager, A.M.; Vats, P.; Kumar-Sinha, C.; Giordano, T.J.; Rainey, W.E.; Else, T. Identification of Somatic Mutations in CLCN2 in Aldosterone-Producing Adenomas. J. Endocr. Soc. 2020, 4, bvaa123. [Google Scholar] [CrossRef]
- Rege, J.; Bandulik, S.; Nanba, K.; Kosmann, C.; Blinder, A.R.; Plain, A.; Vats, P.; Kumar-Sinha, C.; Lerario, A.M.; Else, T.; et al. Somatic SLC30A1 mutations altering zinc transporter ZnT1 cause aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2023, 55, 1623–1631. [Google Scholar] [CrossRef]
- Wu, X.; Azizan, E.A.B.; Goodchild, E.; Garg, S.; Hagiyama, M.; Cabrera, C.P.; Fernandes-Rosa, F.L.; Boulkroun, S.; Kuan, J.L.; Tiang, Z.; et al. Somatic mutations of CADM1 in aldosterone-producing adenomas and gap junction-dependent regulation of aldosterone production. Nat. Genet. 2023, 55, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Rhayem, Y.; Perez-Rivas, L.G.; Dietz, A.; Bathon, K.; Gebhard, C.; Riester, A.; Mauracher, B.; Gomez-Sanchez, C.; Eisenhofer, G.; Schwarzmayr, T.; et al. PRKACA Somatic Mutations Are Rare Findings in Aldosterone-Producing Adenomas. J. Clin. Endocrinol Metab. 2016, 101, 3010–3017. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, Y.; Huang, Z.; Zhu, Y.; Li, L.; Yang, H.; Liang, X.; Qin, Y.; Zhou, J.; Xian, J.; et al. Rare correlation of somatic PRKACA mutations with pregnancy-associated aldosterone- and cortisol-producing adenomas: A case report and literature review. BMC Endocr. Disord. 2024, 24, 116. [Google Scholar] [CrossRef]
- Zhou, J.; Azizan, E.A.B.; Cabrera, C.P.; Fernandes-Rosa, F.L.; Boulkroun, S.; Argentesi, G.; Cottrell, E.; Amar, L.; Wu, X.; O’Toole, S.; et al. Somatic mutations of GNA11 and GNAQ in CTNNB1-mutant aldosterone-producing adenomas presenting in puberty, pregnancy or menopause. Nat. Genet. 2021, 53, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Nanba, K.; Rainey, W.E.; Udager, A.M. Approaches to Gene Mutation Analysis Using Formalin-Fixed Paraffin-Embedded Adrenal Tumor Tissue From Patients With Primary Aldosteronism. Front. Endocrinol. 2021, 12, 683588. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, S.; Yang, H.; Bai, Y.; Shen, Y.; Ren, Y. Immunohistochemistry combined with NGS to assist the differential diagnosis of multiple primary lung cancer with lymph node metastasis: A case report. Front. Oncol. 2023, 13, 1260759. [Google Scholar] [CrossRef]
- Dibb, K.M.; Rose, T.; Makary, S.Y.; Claydon, T.W.; Enkvetchakul, D.; Leach, R.; Nichols, C.G.; Boyett, M.R. Molecular basis of ion selectivity, block, and rectification of the inward rectifier Kir3.1/Kir3.4 K(+) channel. J. Biol. Chem. 2003, 278, 49537–49548. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.Y.; Liao, H.W.; Chueh, J.S.; Pan, C.Y.; Lin, Y.H.; Chen, Y.M.; Chen, P.Y.; Huang, C.L.; Wu, V.C. Pathophysiological and Pharmacological Characteristics of KCNJ5 157-159delITE Somatic Mutation in Aldosterone-Producing Adenomas. Biomedicines 2021, 9, 1026. [Google Scholar] [CrossRef]
- Makara, J.K.; Petheö, G.L.; Tóth, A.; Spät, A. Effect of osmolarity on aldosterone production by rat adrenal glomerulosa cells. Endocrinology 2000, 141, 1705–1710. [Google Scholar] [CrossRef]
- Williams, T.A.; Monticone, S.; Schack, V.R.; Stindl, J.; Burrello, J.; Buffolo, F.; Annaratone, L.; Castellano, I.; Beuschlein, F.; Reincke, M.; et al. Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension 2014, 63, 188–195. [Google Scholar] [CrossRef]
- Tauber, P.; Aichinger, B.; Christ, C.; Stindl, J.; Rhayem, Y.; Beuschlein, F.; Warth, R.; Bandulik, S. Cellular Pathophysiology of an Adrenal Adenoma-Associated Mutant of the Plasma Membrane Ca(2+)-ATPase ATP2B3. Endocrinology 2016, 157, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Rosa, F.L.; Boulkroun, S.; Zennaro, M.C. Genetic and Genomic Mechanisms of Primary Aldosteronism. Trends Mol. Med. 2020, 26, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Monticone, S.; Castellano, I.; Versace, K.; Lucatello, B.; Veglio, F.; Gomez-Sanchez, C.E.; Williams, T.A.; Mulatero, P. Immunohistochemical, genetic and clinical characterization of sporadic aldosterone-producing adenomas. Mol. Cell. Endocrinol. 2015, 411, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Björklund, P.; Lindberg, D.; Akerström, G.; Westin, G. Stabilizing mutation of CTNNB1/beta-catenin and protein accumulation analyzed in a large series of parathyroid tumors of Swedish patients. Mol. Cancer 2008, 7, 53. [Google Scholar] [CrossRef]
- Berthon, A.; Drelon, C.; Ragazzon, B.; Boulkroun, S.; Tissier, F.; Amar, L.; Samson-Couterie, B.; Zennaro, M.C.; Plouin, P.F.; Skah, S.; et al. WNT/β-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum. Mol. Genet. 2014, 23, 889–905. [Google Scholar] [CrossRef]
- O’Hayre, M.; Vázquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. 2013, 13, 412–424. [Google Scholar] [CrossRef]
- Parish, A.J.; Nguyen, V.; Goodman, A.M.; Murugesan, K.; Frampton, G.M.; Kurzrock, R. GNAS, GNAQ, and GNA11 alterations in patients with diverse cancers. Cancer 2018, 124, 4080–4089. [Google Scholar] [CrossRef]
- Tetti, M.; Gong, S.; Veglio, F.; Reincke, M.; Williams, T.A. Primary aldosteronism: Pathophysiological mechanisms of cell death and proliferation. Front. Endocrinol. 2022, 13, 934326. [Google Scholar] [CrossRef]
- Nanba, K.; Blinder, A.R.; Udager, A.M.; Hirokawa, Y.; Miura, T.; Okuno, H.; Moriyoshi, K.; Yamazaki, Y.; Sasano, H.; Yasoda, A.; et al. Double somatic mutations in CTNNB1 and GNA11 in an aldosterone-producing adenoma. Front. Endocrinol. 2024, 15, 1286297. [Google Scholar] [CrossRef]
- Scholl, U.I.; Stölting, G.; Nelson-Williams, C.; Vichot, A.A.; Choi, M.; Loring, E.; Prasad, M.L.; Goh, G.; Carling, T.; Juhlin, C.C.; et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. eLife 2015, 4, e06315. [Google Scholar] [CrossRef]
- Scholl, U.I. CLCN2 clicks with aldosterone-producing adenomas, too! Eur. J. Endocrinol. 2019, 181, C21–C22. [Google Scholar] [CrossRef] [PubMed]
- Mizdrak, M.; Ticinovic Kurir, T.; Mizdrak, I.; Kumric, M.; Krnic, M.; Bozic, J. The Role of the Gap Junction Protein Connexin in Adrenal Gland Tumorigenesis. Int. J. Mol. Sci. 2024, 25, 5399. [Google Scholar] [CrossRef]
- Bell, C.L.; Murray, S.A. Adrenocortical Gap Junctions and Their Functions. Front. Endocrinol. 2016, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.; Ohana, E.; Besser, L.; Hershfinkel, M.; Moran, A.; Sekler, I. A role for ZnT-1 in regulating cellular cation influx. Biochem. Biophys. Res. Commun. 2004, 323, 1145–1150. [Google Scholar] [CrossRef]
- Andrews, G.K.; Wang, H.; Dey, S.K.; Palmiter, R.D. Mouse zinc transporter 1 gene provides an essential function during early embryonic development. Genesis 2004, 40, 74–81. [Google Scholar] [CrossRef]
- Levy, S.; Beharier, O.; Etzion, Y.; Mor, M.; Buzaglo, L.; Shaltiel, L.; Gheber, L.A.; Kahn, J.; Muslin, A.J.; Katz, A.; et al. Molecular basis for zinc transporter 1 action as an endogenous inhibitor of L-type calcium channels. J. Biol. Chem. 2009, 284, 32434–32443. [Google Scholar] [CrossRef]
- Rege, J.; Hoxie, J.; Liu, C.J.; Cash, M.N.; Luther, J.M.; Gellert, L.; Turcu, A.F.; Else, T.; Giordano, T.J.; Udager, A.M.; et al. Targeted Mutational Analysis of Cortisol-Producing Adenomas. J. Clin. Endocrinol. Metab. 2022, 107, e594–e603. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Timmers, H.J.L.M.; Arnaldi, G.; Küsters, B.; Scarpelli, M.; Bathon, K.; Calebiro, D.; Beuschlein, F.; Hermus, A.; Reincke, M. Somatic PRKACA Mutations: Association With Transition From Pituitary-Dependent to Adrenal-Dependent Cushing Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 5651–5657. [Google Scholar] [CrossRef] [PubMed]
- Berthon, A.S.; Szarek, E.; Stratakis, C.A. PRKACA: The catalytic subunit of protein kinase A and adrenocortical tumors. Front. Cell Dev. Biol. 2015, 3, 26. [Google Scholar] [CrossRef]
- Beuschlein, F.; Fassnacht, M.; Assié, G.; Calebiro, D.; Stratakis, C.A.; Osswald, A.; Ronchi, C.L.; Wieland, T.; Sbiera, S.; Faucz, F.R.; et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N. Engl. J. Med. 2014, 370, 1019–1028. [Google Scholar] [CrossRef]
- Goh, G.; Scholl, U.I.; Healy, J.M.; Choi, M.; Prasad, M.L.; Nelson-Williams, C.; Kunstman, J.W.; Korah, R.; Suttorp, A.C.; Dietrich, D.; et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat. Genet. 2014, 46, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.Q.; Stratakis, C.A. How does cAMP/protein kinase A signaling lead to tumors in the adrenal cortex and other tissues? Mol. Cell. Endocrinol. 2011, 336, 162–168. [Google Scholar] [CrossRef]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef]
- Fallo, F.; Castellano, I.; Gomez-Sanchez, C.E.; Rhayem, Y.; Pilon, C.; Vicennati, V.; Santini, D.; Maffeis, V.; Fassina, A.; Mulatero, P.; et al. Histopathological and genetic characterization of aldosterone-producing adenomas with concurrent subclinical cortisol hypersecretion: A case series. Endocrine 2017, 58, 503–512. [Google Scholar] [CrossRef]
- Nanba, K.; Omata, K.; Tomlins, S.A.; Giordano, T.J.; Hammer, G.D.; Rainey, W.E.; Else, T. Double adrenocortical adenomas harboring independent KCNJ5 and PRKACA somatic mutations. Eur. J. Endocrinol. 2016, 175, K1–K6. [Google Scholar] [CrossRef] [PubMed]
- Pitsava, G.; Stratakis, C.A. Genetic Alterations in Benign Adrenal Tumors. Biomedicines 2022, 10, 1041. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, M.C.; Boulkroun, S.; Fernandes-Rosa, F. Genetic Causes of Functional Adrenocortical Adenomas. Endocr. Rev. 2017, 38, 516–537. [Google Scholar] [CrossRef]
- Faconti, L.; McNally, R.J.; Farukh, B.; Adeyemi, O.; Cruickshank, J.K.; Wilkinson, I.B.; Chowienczyk, P.J.; Ojji, D. Differences in hypertension phenotypes between Africans and Europeans: Role of environment. J. Hypertens. 2020, 38, 1278–1285. [Google Scholar] [CrossRef]
- Li, X.T. The modulation of potassium channels by estrogens facilitates neuroprotection. Front. Cell Dev. Biol. 2022, 10, 998009. [Google Scholar] [CrossRef]
- Gannon, A.L.; O’Hara, L.; Mason, J.I.; Jørgensen, A.; Frederiksen, H.; Milne, L.; Smith, S.; Mitchell, R.T.; Smith, L.B. Androgen receptor signalling in the male adrenal facilitates X-zone regression, cell turnover and protects against adrenal degeneration during ageing. Sci. Rep. 2019, 9, 10457. [Google Scholar] [CrossRef]
- Tissier, F.; Cavard, C.; Groussin, L.; Perlemoine, K.; Fumey, G.; Hagneré, A.M.; René-Corail, F.; Jullian, E.; Gicquel, C.; Bertagna, X.; et al. Mutations of beta-catenin in adrenocortical tumors: Activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005, 65, 7622–7627. [Google Scholar] [CrossRef]
- Tseng, C.S.; Chan, C.K.; Lee, H.Y.; Pan, C.T.; Peng, K.Y.; Wang, S.M.; Huang, K.H.; Tsai, Y.C.; Wu, V.C.; Chueh, J.S.; et al. Treatment of primary aldosteronism: Clinical practice guidelines of the Taiwan Society of Aldosteronism. J. Formos. Med. Assoc. Taiwan Yi Zhi 2024, 123, S125–S134. [Google Scholar] [CrossRef] [PubMed]
- Lenzini, L.; Rossitto, G.; Maiolino, G.; Letizia, C.; Funder, J.W.; Rossi, G.P. A Meta-Analysis of Somatic KCNJ5 K(+) Channel Mutations In 1636 Patients With an Aldosterone-Producing Adenoma. J. Clin. Endocrinol. Metabol. 2015, 100, E1089–E1095. [Google Scholar] [CrossRef] [PubMed]
- Vilela, L.A.P.; Rassi-Cruz, M.; Guimaraes, A.G.; Moises, C.C.S.; Freitas, T.C.; Alencar, N.P.; Petenuci, J.; Goldbaum, T.S.; Maciel, A.A.W.; Pereira, M.A.A.; et al. KCNJ5 Somatic Mutation Is a Predictor of Hypertension Remission After Adrenalectomy for Unilateral Primary Aldosteronism. J. Clin. Endocrin. Metabol. 2019, 104, 4695–4702. [Google Scholar] [CrossRef]
- Kato, H.; Kitamoto, T.; Kimura, S.; Sunouchi, T.; Hoshino, Y.; Hidaka, N.; Tsurutani, Y.; Ito, N.; Makita, N.; Nishikawa, T.; et al. Cardiovascular Outcomes of KCNJ5 Mutated Aldosterone-Producing Adenoma: A Systematic Review. Endocr. Pract. 2024, 30, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wu, L.; Jiang, L.; Su, T.; Zhou, W.; Zhong, X.; Xie, J.; Sun, F.; Zhu, Y.; Jiang, Y.; et al. KCNJ5 Mutation Contributes to Complete Clinical Success in Aldosterone-Producing Adenoma: A Study From a Single Center. Endocr. Pract. 2021, 27, 736–742. [Google Scholar] [CrossRef]
- Williams, T.A.; Peitzsch, M.; Dietz, A.S.; Dekkers, T.; Bidlingmaier, M.; Riester, A.; Treitl, M.; Rhayem, Y.; Beuschlein, F.; Lenders, J.W.; et al. Genotype-Specific Steroid Profiles Associated With Aldosterone-Producing Adenomas. Hypertension 2016, 67, 139–145. [Google Scholar] [CrossRef]
- Dekkers, T.; ter Meer, M.; Lenders, J.W.; Hermus, A.R.; Schultze Kool, L.; Langenhuijsen, J.F.; Nishimoto, K.; Ogishima, T.; Mukai, K.; Azizan, E.A.; et al. Adrenal nodularity and somatic mutations in primary aldosteronism: One node is the culprit? J. Clin. Endocrinol. Metab. 2014, 99, E1341–E1351. [Google Scholar] [CrossRef]
- Scholl, U.I.; Healy, J.M.; Thiel, A.; Fonseca, A.L.; Brown, T.C.; Kunstman, J.W.; Horne, M.J.; Dietrich, D.; Riemer, J.; Kücükköylü, S.; et al. Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin. Endocrinol. 2015, 83, 779–789. [Google Scholar] [CrossRef]
- Rege, J.; Turcu, A.F.; Rainey, W.E. Primary aldosteronism diagnostics: KCNJ5 mutations and hybrid steroid synthesis in aldosterone-producing adenomas. Gland Surg. 2020, 9, 3–13. [Google Scholar] [CrossRef]
- Aiga, K.; Kometani, M.; Aono, D.; Yoneda, T. Recurrence of Primary Aldosteronism After Surgery in Aldosterone-producing Adenoma With KCNJ5 Gene Mutation. JCEM Case Rep. 2023, 1, luac032. [Google Scholar] [CrossRef]
- Scholl, U.I.; Abriola, L.; Zhang, C.; Reimer, E.N.; Plummer, M.; Kazmierczak, B.I.; Zhang, J.; Hoyer, D.; Merkel, J.S.; Wang, W.; et al. Macrolides selectively inhibit mutant KCNJ5 potassium channels that cause aldosterone-producing adenoma. J. Clin. Investig. 2017, 127, 2739–2750. [Google Scholar] [CrossRef]
- Wu, X.; Senanayake, R.; Goodchild, E.; Bashari, W.A.; Salsbury, J.; Cabrera, C.P.; Argentesi, G.; O’Toole, S.M.; Matson, M.; Koo, B.; et al. [11C]metomidate PET-CT versus adrenal vein sampling for diagnosing surgically curable primary aldosteronism: A prospective, within-patient trial. Nat. Med. 2023, 29, 190–202. [Google Scholar] [CrossRef]
- Hacini, I.; De Sousa, K.; Boulkroun, S.; Meatchi, T.; Amar, L.; Zennaro, M.C.; Fernandes-Rosa, F.L. Somatic mutations in adrenals from patients with primary aldosteronism not cured after adrenalectomy suggest common pathogenic mechanisms between unilateral and bilateral disease. Eur. J. Endocrinol. 2021, 185, 405–412. [Google Scholar] [CrossRef]
- Turcu, A.F.; Tezuka, Y.; Lim, J.S.; Salman, Z.; Sehgal, K.; Liu, H.; Larose, S.; Parksook, W.W.; Williams, T.A.; Cohen, D.L.; et al. Multifocal, Asymmetric Bilateral Primary Aldosteronism Cannot be Excluded by Strong Adrenal Vein Sampling Lateralization: An International Retrospective Cohort Study. Hypertension 2024, 81, 604–613. [Google Scholar] [CrossRef]
- Tetti, M.; Brüdgam, D.; Burrello, J.; Udager, A.M.; Riester, A.; Knösel, T.; Beuschlein, F.; Rainey, W.E.; Reincke, M.; Williams, T.A. Unilateral Primary Aldosteronism: Long-Term Disease Recurrence After Adrenalectomy. Hypertension 2024, 81, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Pauzi, F.A.; Azizan, E.A. Functional Characteristic and Significance of Aldosterone-Producing Cell Clusters in Primary Aldosteronism and Age-Related Hypertension. Front. Endocrinol. 2021, 12, 631848. [Google Scholar] [CrossRef]
- Xiang, H.; Zhang, T.; Song, W.; Yang, D.; Zhu, X. Adrenalectomy for primary aldosteronism and its related surgical characteristics. Front. Endocrinol. 2024, 15, 1416287. [Google Scholar] [CrossRef]
- Nishimoto, K.; Tomlins, S.A.; Kuick, R.; Cani, A.K.; Giordano, T.J.; Hovelson, D.H.; Liu, C.J.; Sanjanwala, A.R.; Edwards, M.A.; Gomez-Sanchez, C.E.; et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc. Natl. Acad. Sci. USA 2015, 112, E4591–E4599. [Google Scholar] [CrossRef]
| Gene | Frequency in Europeans | Frequency in East-Asians | Frequency in Africans | References |
|---|---|---|---|---|
| KCNJ5 mutations | 590/1486 (39.7%) | 526/784 (67.1%) | 25/73 (34.2%) | |
| p.Gly151Arg; p.Leu168Arg | 8/22 (36.4%) | - | - | [11] |
| p.Gly151Arg; p.Leu168Arg | 129/380 (33.9%) | - | - | [12] |
| p.Glu145Gln; p.Gly151Arg; p.Leu168Arg | 157/348 (45.1%) | - | - | [22] |
| p.Gly151Arg; p.Leu168Arg; p.Ile157del | 4/8 (50.0%) | - | - | [23] |
| p.Gly151Arg; p.Leu168Arg | - | 15/23 (65.2%) | - | [25] |
| p.Gly151Arg; p.Leu168Arg | 10/28 (35.7%) | - | - | [26] |
| p.Trp126Arg; p.Gly151Arg; p.Thr158Ala; p.Leu168Arg | 180/474 (38.0%) | - | - | [28] |
| p.Arg115Trp; p.Glu145Gln; p.Gly151Arg; p.Leu168Arg; p.Glu246Gly | - | 26/69 (42.0%) | - | [29] |
| p.Glu145Gln; p.Gly151Arg; p.Ile157del; p.Thr158Ala; p.Leu168Arg | - | 75/108 (69.4%) | - | [30] |
| p.Gly151Arg; p.Ile157del; p.Thr158Ala; p.Leu168Arg | - | 88/91 (96.7%) | - | [31] |
| p.Thr148_Thr149insArg; p.Gly151Arg; p.Thr158Ala; p.Leu168Arg | - | 129/168 (76.8%) | - | [32] |
| NA | 92/198 (46.5%) | - | - | [33] |
| p.Glu145Lys; p.Gly151Arg; p.Leu168Arg | 10/28 (35.7%) | - | - | [34] |
| p.Gly151Arg; p.Ile157del; p.Thr158Ala; p.Leu168Arg | - | 116/219 (53.0%) | - | [35] |
| p.[Thr148Ile;Thr149Ser]; p.Thr149delinsThrThr; p.Thr149delinsMetAla; p.Gly151Arg; p.Leu168Arg | - | - | 25/73 (34.2%) | [36] |
| p.Glu145Gln; p.Gly151Arg; p.Thr158Ala; p.Leu168Arg | - | 77/106 (72.6%) | - | [37] |
| CACNA1D mutations | 57/682 (8.4%) | 16/274 (5.8%) | 32/75 (42.7%) | |
| pVal401Leu; p.Gly403Arg; p.Phe747Leu; p.Val1353Met | 5/165 (3.0%) | - | - | [15] |
| p.Gly403Arg; p.Ile770Met | 4/15 (26.7%) | - | 1/2 (50.0%) | [27] |
| p.Val259Asp; p.Gly403Arg; p.Ser652Leu; p.Leu655Pro; p.Tyr741Cys; p.Phe747Val; p.Phe747Leu; p.Ile750Met; p.Ile750Phe; p.Val979Asp; p.Val981Asn; p.Ala998Ile; p.Ala998Val; p.Val1151Phe; p.Ile1152Asn; p.Pro1336Arg; p.Val1338Met; p.Met1354Ile | 44/474 (9.3%) | - | - | [28] |
| p.Gly403Arg | - | 1/168 (0.6%) | - | [32] |
| p.Gly403Arg; p.Phe747Leu; p.Arg990His; p.Val1153Gly | 4/28 (14.3%) | - | - | [34] |
| p.Val309Ala; p.Val401Leu; p.Gly403Arg; p.Arg619Pro; p.Ser652Leu; p.Phe747Val/Leu/Cys; p.Ile750Phe/Met; p.Arg990Gly; p.Arg993Thr; p.Ala998Val; p.Cys1007Arg; p.Ile1015Ser; p.Val1151Phe | - | - | 31/73 (42.5%) | [36] |
| p.Gly403Arg; p.Ser652Leu; p.Phe747Val; p.Ser969Leu; p.Arg990His; p.Ala998Val/Ile; p.Ile1015Thr; p.Val1338Met | - | 15/106 (14.1%) | - | [37] |
| ATP1A1 mutations | 42/667 (6.3%) | 11/365 (3.0%) | 6/73 (8.2%) | |
| p.Met102_Leu103del; p.Met102_Ile106del; p.Leu103_Leu104del; p.Leu104Arg; p.Phe956_Glu961del; p.Phe959_Glu961del; p.Glu960_Leu964del | 10/165 (6.1%) | - | - | [15] |
| p.Gly99Arg; p.Phe100_Leu104del; p.Leu104Arg; p.Val332Gly | 25/474 (5.3%) | - | - | [28] |
| p.Leu104Arg | - | 2/91 (2.2%) | - | [31] |
| p.Met102_Leu103del; p.Leu104Arg | - | 4/168 (2.4%) | - | [32] |
| p.Phe100_Leu104del; p.Leu104Arg | 7/28 (25.0%) | [34] | ||
| p.Leu104Arg; p.Ile955_Glu960delinsLys | - | - | 6/73 (8.2%) | [36] |
| p.Leu104Arg; p.Phe959_Glu961delinsLeu; p.Glu960_Ala965delinsAlaLeuVal | - | 5/106 (4.7%) | - | [37] |
| ATP2B3 mutations | 11/639 (1.7%) | 6/365 (1.6%) | 3/73 (4.1%) | |
| p.Thr423_Leu425del; p.Val424_Leu425del; p.Val424_Val426del; p.Val426_Val429del | 5/165 (3.0%) | - | - | [15] |
| p.Leu424_Val425del; p.Leu425_Val426del; p.Val426_Val427del | 8/474 (1.7%) | - | - | [28] |
| p.Tyr410Asp | - | 1/91 (1.1%) | - | [31] |
| p.Val422-Val426delinsSerThrLeu | - | 1/168 (0.6%) | - | [32] |
| p.Val424_Leu425del | - | - | 3/73 (4.1%) | [36] |
| p.Val424_Leu425del; p.Leu425_Val426del | - | 4/106 (3.8%) | - | [37] |
| CTNNB1 mutations | 10/198 (5.1%) | 8/219 (3.6%) | 0 | |
| p.Thr41Ala; p.Ser45Phe; p.Ser45Pro | 10/198 (5.1%) | - | - | [33] |
| p.Ser45Phe; p.Ser45Pro | - | 8/219 (3.6%) | - | [35] |
| GNA11 mutations (co-occurring with CTNNB1) | 11/27 (40.7%) | 0 | 0 | |
| p.Gln209Pro with p.Gly34Arg/p.Ser45Phe/p.Ser45Pro; p.Gln209His with p.Ser33Cys/p.Thr41Ala/p.Ser45Phe | 11/27 (40.7%) | - | - | [46] |
| GNAQ mutations (co-occurring with CTNNB1) | 5/27 (18.5%) | 0 | 0 | |
| p.Gln209His with p.Gly34Glu; p.Gln209Leu with p.Gly34Arg | 5/27 (18.5%) | - | - | [46] |
| CACNA1H mutations | 8/115 (7.0%) | 0 | 0 | |
| p.Ile1430Thr | 3/75 (4.0%) | - | - | [38] |
| CLCN2 mutations | 4/207 (1.9%) | 0 | 0 | |
| p.Gly24Asp | 1/12 | - | - | [39] |
| p.Gly24Asp | 1/80 | - | - | [40] |
| p.Gly24Asp; c.64-2_74del | 2/115 | - | - | [41] |
| CADM1 mutations | 6/40 (15%) | 0 | 0 | |
| p.Gly379Asp; p.Val380Asp | 6/40 (15%) | - | - | [43] |
| SLC30A1 mutations | 3/118 (2.5%) | 2/68 (2.9%) | 0 | |
| p.Leu49_Leu55del; p.Leu51_Ala57del | 3/118 (2.5%) | 2/68 (2.9%) | - | [42] |
| PRKACA mutations | 1/122 (0.8%) | 1/20 (5%) | 1/122 (0.8%) | |
| p.His88Asp; p.Leu206Arg | 1/122 (0.8%) | - | 1/122 (0.8%) | [44] |
| p.Leu206Arg | - | 1/20 (5%) | - | [45] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abd Rahman, A.N.; Azizan, E.A. Somatic Mutations Associated with Aldosterone-Producing Adenomas (APAs). Genes 2025, 16, 778. https://doi.org/10.3390/genes16070778
Abd Rahman AN, Azizan EA. Somatic Mutations Associated with Aldosterone-Producing Adenomas (APAs). Genes. 2025; 16(7):778. https://doi.org/10.3390/genes16070778
Chicago/Turabian StyleAbd Rahman, Aina Nadheera, and Elena Aisha Azizan. 2025. "Somatic Mutations Associated with Aldosterone-Producing Adenomas (APAs)" Genes 16, no. 7: 778. https://doi.org/10.3390/genes16070778
APA StyleAbd Rahman, A. N., & Azizan, E. A. (2025). Somatic Mutations Associated with Aldosterone-Producing Adenomas (APAs). Genes, 16(7), 778. https://doi.org/10.3390/genes16070778