
Brogidirsen and Exon 44 Skipping for Duchenne Muscular Dystrophy: Advances and Challenges in RNA-Based Therapy



Abstract
1. Introduction to Duchenne Muscular Dystrophy and Treatments
1.1. Duchenne Muscular Dystrophy (DMD) Pathology
1.2. Antisense DMD Treatments
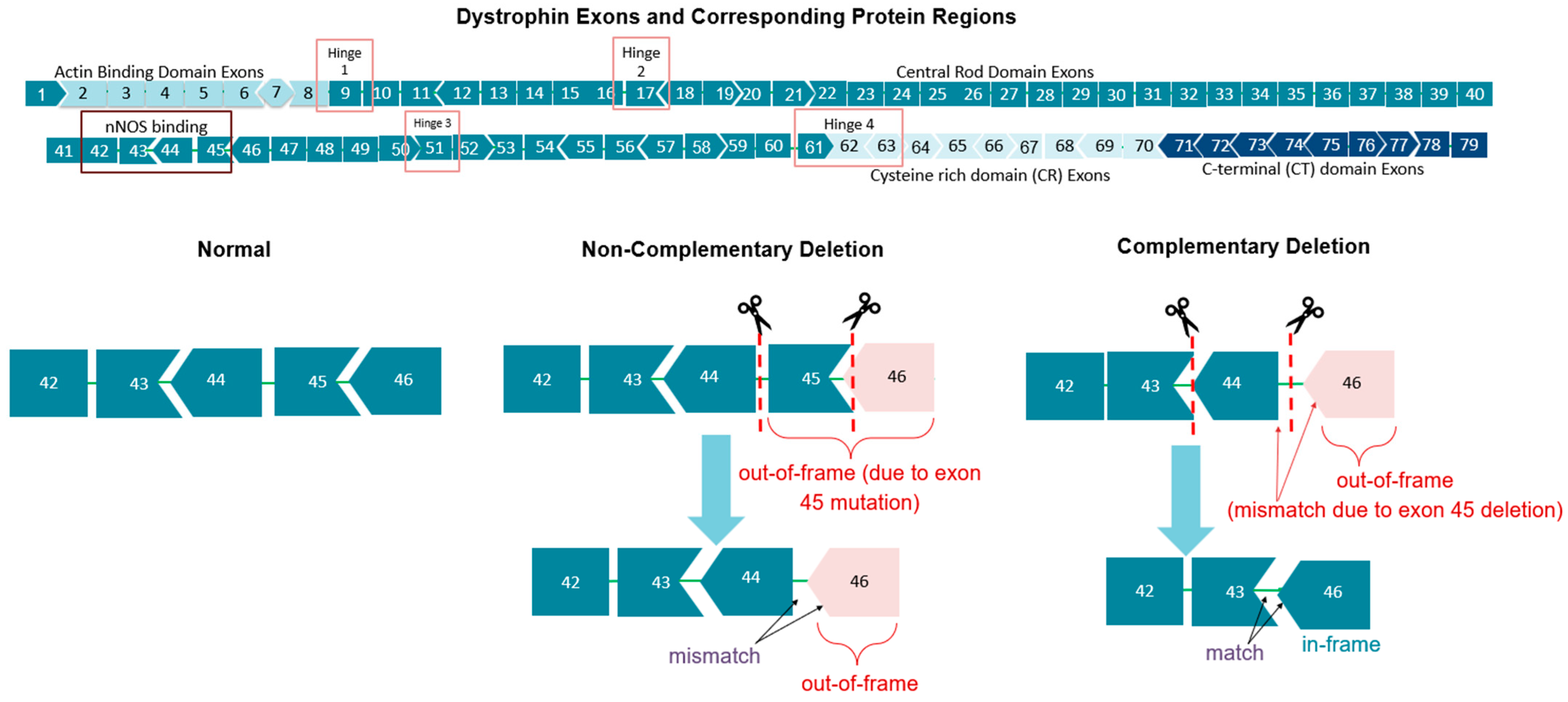
2. Exon 44-Skipping Therapies
2.1. Mechanism and Targets
2.2. Current Pre-Clinical and Clinical Trial Results for AOC1044 and ENTR-601-44
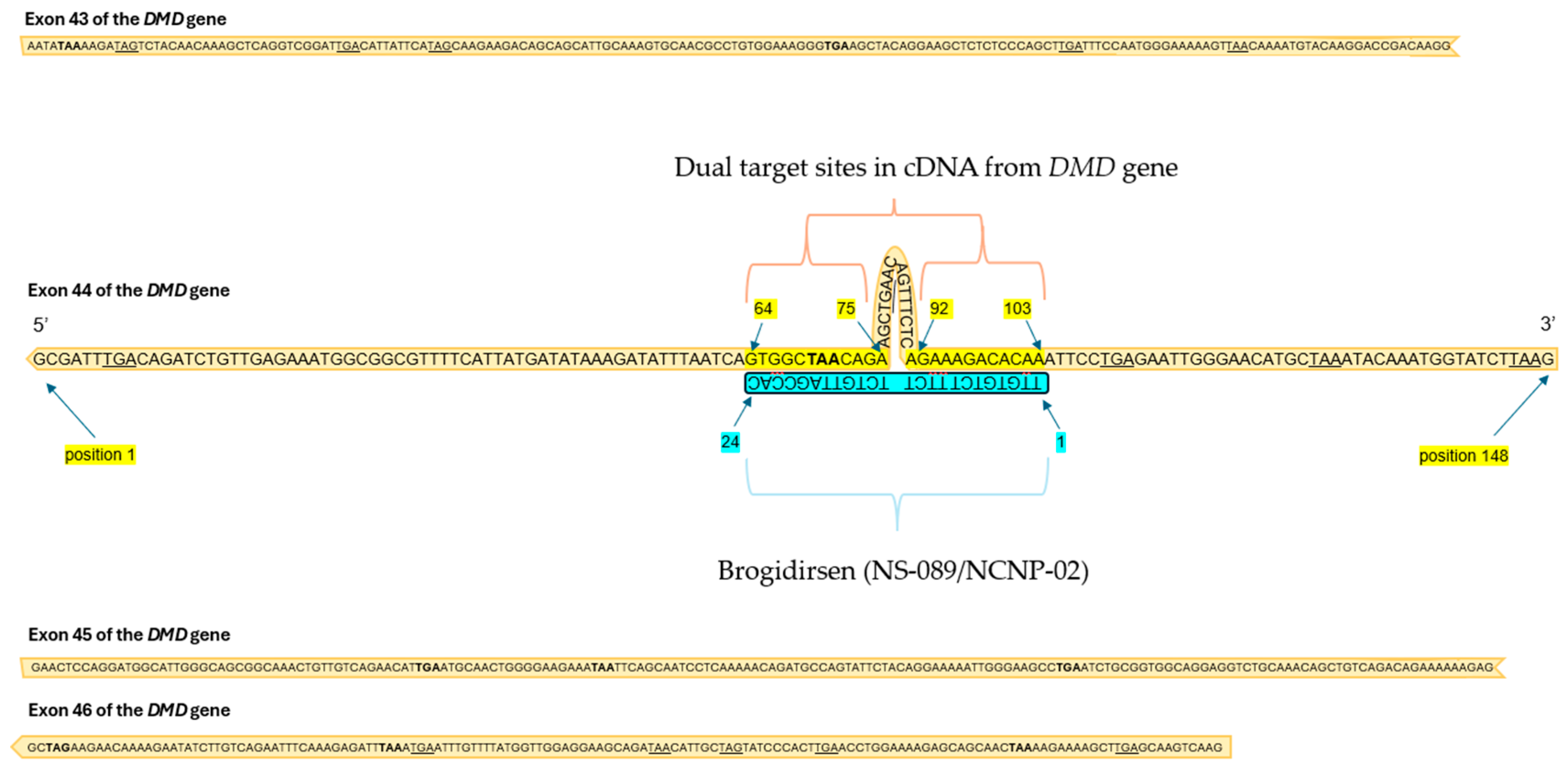
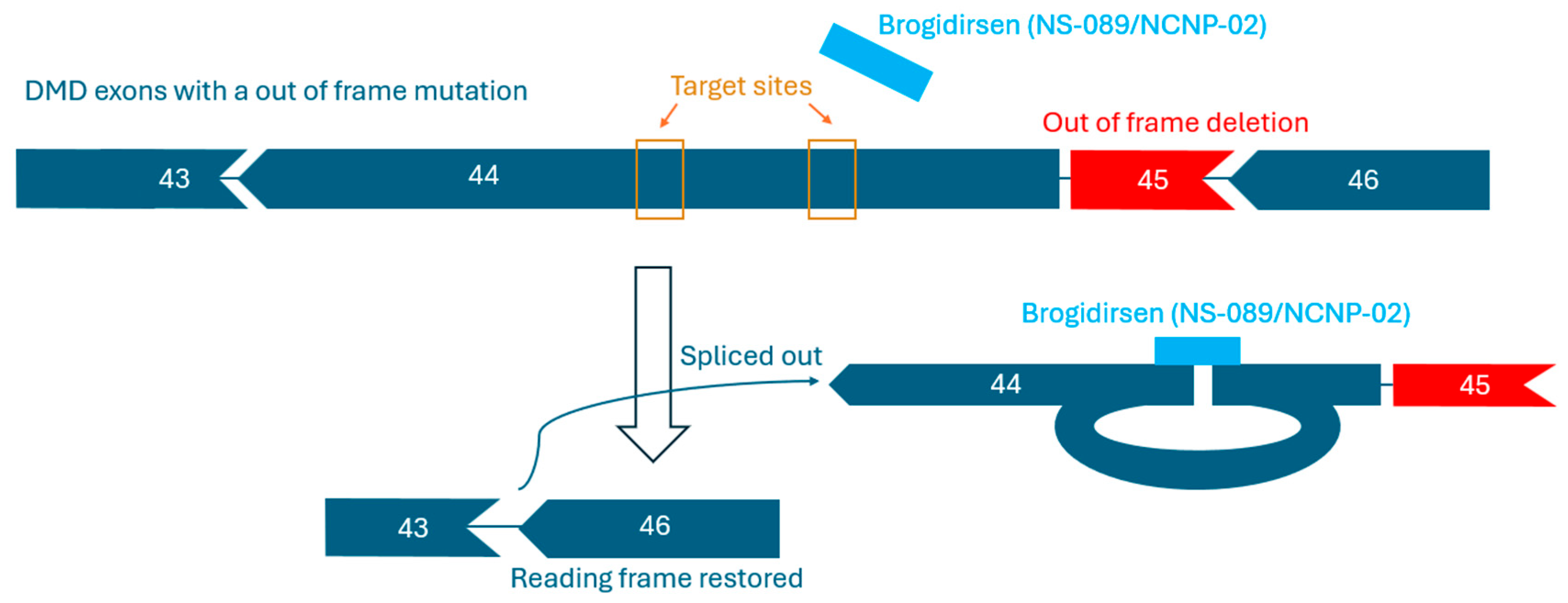
3. Brogidirsen Studies and Trials
3.1. Preclinical Study Results
3.2. Clinical Findings and Pharmacology
4. Comparison of Brogidirsen to Other Exon 44-Targeting Therapies
5. Addressing Challenges Related to Exon-Skipping Therapies
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| DMD | Duchenne muscular dystrophy |
| ASO | Antisense oligonucleotide |
| PMO | Phosphorodiamidate morpholino oligomer |
| EEV | Endosomal Escape Vehicle |
| MTD | Maximum tolerated dose |
| TfR1 | Transferrin receptor 1 |
| BMD | Becker muscular dystrophy |
References
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifiro, G. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.Q.; McNally, E.M. The dystrophin complex: Structure, function, and implications for therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Salari, N.; Fatahi, B.; Valipour, E.; Kazeminina, M.; Fatahian, R.; Kiaei, A.; Shohaimi, S.; Mohammadi, M. Global prevalence of Duchenne and Becker muscular dystrophy: A systematic review and meta-analysis. J. Orthop. Surg. Res. 2022, 17, 96. [Google Scholar] [CrossRef]
- Sussman, M. Duchenne Muscular Dystrophy. J. Am. Acad. Orthop. Surg. 2002, 10, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef]
- Shieh, P.B. Emerging Strategies in the Treatment of Duchenne Muscular Dystrophy. Neurotherapeutics 2018, 15, 840–848. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Review Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; Kock, S.D.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Liu, D.; Ahmet, A.; Ward, L.; Krishnamoorthy, P.; Mandelcorn, E.D.; Leigh, R.; Brown, J.P.; Cohen, A.; Kim, H. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin. Immunol. 2013, 9, 30. [Google Scholar] [CrossRef]
- Gatto, F.; Benemei, S.; Piluso, G.; Bello, L. The complex landscape of DMD mutations: Moving towards personalized medicine. Front. Genet. 2024, 15, 1360224. [Google Scholar] [CrossRef]
- Godfrey, C.; Muses, S.; McClorey, G.; Wells, K.E.; Coursindel, T.; Terry, R.L.; Betts, C.; Hammond, S.; O’Donovan, L.; Hildyard, J.C.W.; et al. How much dystrophin is enough: The physiological consequences of different levels of dystrophin in the mdx mouse. Hum. Mol. Genet. 2015, 24, 4225–4237. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Conway, K.M.; Fapo, O.; Holtzer, C.; Meany, F.J.; Andrews, J. Time to diagnosis of Duchenne muscular dystrophy remains unchanged: Findings from the Muscular Dystrophy Surveillance, Tracking, and Research Network, 2000–2015. Muscle Nerve 2022, 66, 193–197. [Google Scholar] [CrossRef]
- Abbs, S.; Tuffery-Giraud, S.; Bakker, E.; Ferlini, A.; Sejersen, T.; Mueller, C.R. Best Practice Guidelines on molecular diagnostics in Duchenne/Becker muscular dystrophies. Neuromuscul. Disord. 2010, 20, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.; Duval, B.; Bouffard, G. Evidence-based path to newborn screening for duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef]
- Emery, A.E. Clinical and molecular studies in Duchenne muscular dystrophy. Prog. Clin. Biol. Res. 1989, 306, 15–28. [Google Scholar]
- Andrews, J.G.; Wahl, R.A. Duchenne and Becker muscular dystrophy in adolescents: Current perspectives. Adolesc. Health Med. Ther. 2018, 9, 53–63. [Google Scholar] [CrossRef]
- Zambon, A.A.; Ayyar Gupta, V.; Ridout, D.; Manzur, A.Y.; Baranello, G.; Trucco, F.; Muntoni, F.; Tirupath, S.; Douglas, M.; McFetridge, J.; et al. Peak functional ability and age at loss of ambulation in Duchenne muscular dystrophy. Dev. Med. Child Neurol. 2022, 64, 979–988. [Google Scholar] [CrossRef]
- Garg, S. Management of scoliosis in patients with Duchenne muscular dystrophy and spinal muscular atrophy: A literature review. J. Pediatr. Rehabil. Med. 2016, 9, 23–29. [Google Scholar] [CrossRef]
- Hendriksen, J.G.M.; Vles, J.S.H. Neuropsychiatric disorders in males with duchenne muscular dystrophy: Frequency rate of attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder, and obsessive--compulsive disorder. J. Child Neurol. 2008, 23, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Banihani, R.; Smile, S.; Yoon, G.; Dupuis, A.; Mosleh, M.; Snider, A.; McAdam, L. Cognitive and Neurobehavioral Profile in Boys With Duchenne Muscular Dystrophy. J. Child Neurol. 2015, 30, 1472–1482. [Google Scholar] [CrossRef]
- Houwen-van Opstal, S.L.S.; Heutinck, L.; Jansen, M.; Krom, Y.D.; Cup, E.H.C.; Hendriksen, J.G.M.; Willemsen, M.A.A.P.; Verschuuren, J.J.G.M.; Niks, E.H.; de Groot, I.J.M. Occurrence of symptoms in different stages of Duchenne muscular dystrophy and their impact on social participation. Muscle Nerve 2021, 64, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.R.; Zhang, Y.; McDermott, S.; Wang, Y.; Cai, B.; Conway, K.M.; Paramsothy, P.; Royer, J.; Venkatesh, S.; Howard, J.F., Jr.; et al. Racial and Ethnic Differences in Timing of Diagnosis and Clinical Services Received in Duchenne Muscular Dystrophy. Neuroepidemiology 2023, 57, 90–99. [Google Scholar] [CrossRef]
- Posner, N.; Manjelievskaia, J.; Talaga, A.K.; Richards, M.; Lew, C.R.; Merla, V.; Jimenez Alvir, J.M.; Nelson, S.F. Real-world treatment and health care utilization among patients with Duchenne muscular dystrophy by race and ethnicity in a Medicaid population. J. Manag. Care Spec. Pharm. 2025, 31, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, O.; Yokota, T. Developing DMD therapeutics: A review of the effectiveness of small molecules, stop-codon readthrough, dystrophin gene replacement, and exon-skipping therapies. Expert Opin. Investig. Drugs 2021, 30, 167–176. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef]
- Anwar, S.; Yokota, T. Golodirsen for Duchenne muscular dystrophy. Drugs Today 2020, 56, 491. [Google Scholar] [CrossRef]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Clemens, P.R.; Previtera, M.L.; Crozier, R.A.; Magnus, L.; Hoffman, E.; Komaki, H.; Aoki, Y.; Rao, V.K. Brogidirsen, an Investigational exon 44 Skipping Agent for the Treatment of Duchenne Muscular Dystrophy: Clinical Trial Design (Phase 2); Muscular Dystrophy Association National Office: Chicago, IL, USA, 2024. [Google Scholar]
- Adkin, C.F.; Meloni, P.L.; Fletcher, S.; Adams, A.M.; Muntoni, F.; Wong, B.; Wilton, S.D. Multiple exon skipping strategies to by-pass dystrophin mutations. Neuromuscul. Disord. 2012, 22, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Findlay, A.R.; Wein, N.; Kaminoh, Y.; Taylor, L.E.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann. Neurol. 2015, 77, 668–674. [Google Scholar] [CrossRef]
- Anwar, S.; Yokota, T. The Dysferlinopathies Conundrum: Clinical Spectra, Disease Mechanism and Genetic Approaches for Treatments. Biomolecules 2024, 14, 256. [Google Scholar] [CrossRef] [PubMed]
- Tsoumpra, M.K.; Fukumoto, S.; Matsumoto, T.; Takeda, S.; Wood, M.J.A.; Aoki, Y. Peptide-conjugate antisense based splice-correction for Duchenne muscular dystrophy and other neuromuscular diseases. EBioMedicine 2019, 45, 630–645. [Google Scholar] [CrossRef]
- Echigoya, Y.; Lim, K.R.Q.; Melo, D.; Bao, B.; Trieu, N.; Mizobe, Y.; Maruyama, R.; Mamchaoui, K.; Tanihata, J.; Aoki, Y.; et al. Exons 45–55 Skipping Using Mutation-Tailored Cocktails of Antisense Morpholinos in the DMD Gene. Mol. Ther. 2019, 27, 2005–2017. [Google Scholar] [CrossRef]
- Min, Y.-L.; Chemello, F.; Li, H.; Rodriguez-Caycedo, C.; Sánchez-Ortiz, E.; Mireault, A.A.; McAnally, J.R.; Shelton, J.M.; Zhang, Y.; Bassel-Duby, R.; et al. Correction of Three Prominent Mutations in Mouse and Human Models of Duchenne Muscular Dystrophy by Single-Cut Genome Editing. Mol. Ther. 2020, 28, 2044–2055. [Google Scholar] [CrossRef]
- Nakamura, A.; Fueki, N.; Shiba, N.; Motoki, H.; Miyazaki, D.; Nishizawa, H.; Echigoya, Y.; Yokota, T.; Aoki, Y.; Takeda, S.I. Deletion of exons 3−9 encompassing a mutational hot spot in the DMD gene presents an asymptomatic phenotype, indicating a target region for multiexon skipping therapy. J. Hum. Genet. 2016, 61, 663–667. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; van Deutekom, J.C.; Heemskerk, H.; t’Hoen, P.; de Kimpe, S. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Petersen, M.; Nielsen, C.B.; Nielsen, K.E.; Jensen, G.A.; Bondensgaard, K.; Singh, S.K.; Rajwanshi, V.K.; Koshkin, A.A.; Dahl, B.M.; Wengel, J. The conformations of locked nucleic acids (LNA). J. Mol. Recognit. 2000, 13, 44–53. [Google Scholar] [CrossRef]
- Hill, A.C.; Hall, J. The MOE Modification of RNA: Origins and Widescale Impact on the Oligonucleotide Therapeutics Field. Helv. Chim. Acta 2023, 106, e202200169. [Google Scholar] [CrossRef]
- Sabrina Haque, U.; Kohut, M.; Yokota, T. Comprehensive review of adverse reactions and toxicology in ASO-based therapies for Duchenne Muscular Dystrophy: From FDA-approved drugs to peptide-conjugated ASO. Curr. Res. Toxicol. 2024, 7, 100182. [Google Scholar] [CrossRef]
- Devi, G.R. Delivery of Phosphorodiamidate Morpholino Antisense Oligomers in Cancer Cells. Methods Mol. Biol. 2009, 542, 351–361. [Google Scholar] [PubMed]
- Summerton, J.; Weller, D. Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Maksudov, F.; Kliuchnikov, E.; Pierson, D.; Ujwal, M.L.; Marx, K.A.; Chanda, A.; Barsegov, V. Therapeutic phosphorodiamidate morpholino oligonucleotides: Physical properties, solution structures, and folding thermodynamics. Mol. Ther. Nucleic Acids 2023, 31, 631–647. [Google Scholar] [CrossRef] [PubMed]
- Hudziak, R.M.; Barofsky, E.; Barofsky, D.F.; Weller, D.L.; Huang, S.B.; Weller, D.D. Resistance of Morpholino Phosphorodiamidate Oligomers to Enzymatic Degradation. Antisense Nucleic Acid Drug Dev. 1996, 6, 267–272. [Google Scholar] [CrossRef]
- Moulton, J.D.; Jiang, S. Gene Knockdowns in Adult Animals: PPMOs and Vivo-Morpholinos. Molecules 2009, 14, 1304–1323. [Google Scholar] [CrossRef]
- Palacio-Castañeda, V.; Brock, R.; Verdurmen, W.P.R. Generation of Protein-Phosphorodiamidate Morpholino Oligomer Conjugates for Efficient Cellular Delivery via Anthrax Protective Antigen. Methods Mol. Biol. 2022, 2434, 129–141. [Google Scholar]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.; Yokota, T. Is Duchenne gene therapy a suitable treatment despite its immunogenic class effect? Expert Opin. Drug Saf. 2025, 24, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Ersöz, E.; Demir-Dora, D. Unveiling the potential of antisense oligonucleotides: Mechanisms, therapies, and safety insights. Drug Dev. Res. 2024, 85, e22187. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of More than 7,000 Duchenne Muscular Dystrophy Mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Stahl, M.; Zhu, Y.; Goel, V.; Leung, L.; Tami, Y.; Hardin, T.; Etxaniz, U.; Kovach, P.; Herzog, J.; Hughes, S.; et al. AOC 1044 as a Novel Therapeutic Approach for DMD Patients Amenable to Exon 44 Skipping: EXPLORE44TM Phase 1/2 Healthy Volunteer Data; Muscular Dystrophy Association National Office: Orlando, FL, USA, 2024. Available online: https://clinicaltrials.gov/ct2/show/NCT05670730 (accessed on 16 June 2025).
- Cochran, M.; Marks, I.; Albin, T.; Aras, D.; Kovach, P.; Darimont, B.; Huang, H.; Etxaniz, U.; Kwon, H.W.; Shi, Y.; et al. Structure–Activity Relationship of Antibody–Oligonucleotide Conjugates: Evaluating Bioconjugation Strategies for Antibody–Phosphorodiamidate Morpholino Oligomer Conjugates for Drug Development. J. Med. Chem. 2024, 67, 14868–14884. [Google Scholar] [CrossRef]
- Wilton-Clark, H.; Yokota, T. Recent Trends in Antisense Therapies for Duchenne Muscular Dystrophy. Pharmaceutics 2023, 15, 778. [Google Scholar] [CrossRef]
- Etxaniz, U.; Marks, I.; Albin, T.; Diaz, M.; Bhardwaj, R.; Anderson, A.; Tyaglo, O.; Hoang, T.; Missinato, M.A.; Svensson, K.; et al. AOC 1044 induces exon 44 skipping and restores dystrophin protein in preclinical models of Duchenne muscular dystrophy. Nucleic Acids Res. 2025, 53, gkaf241. [Google Scholar] [CrossRef]
- Nguyen, Q.; Yokota, T. Antisense oligonucleotides for the treatment of cardiomyopathy in Duchenne muscular dystrophy. Am. J. Transl. Res. 2019, 11, 1202–1218. [Google Scholar]
- Entrada Therapeutics. Entrada Therapeutics Announces FDA Removal of Clinical Hold on ENTR-601-44. 2025. Available online: www.entradatx.com (accessed on 16 June 2025).
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Cross, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef]
- Sahni, A.; Qian, Z.; Pei, D. Cell-Penetrating Peptides Escape the Endosome by Inducing Vesicle Budding and Collapse. ACS Chem. Biol. 2020, 15, 2485–2492. [Google Scholar] [CrossRef]
- Oldham, M.; Estrella, N.; Kumar, A.; Hicks, A.; Brennan, C.; Blake, S.; Li, X.; Pathak, A.; Kheirabadi, M.; Dougherty, P.; et al. 433P Therapeutic potential of ENTR-601-44, an Endosomal Escape Vehicle (EEVTM)—Oligonucleotide Conjugate for the treatment of exon 44 skip amenable DMD. Neuromuscul. Disord. 2024, 43, 104441.304. [Google Scholar] [CrossRef]
- Wahab, E. A Study to Investigate How ENTR-601-44 Behaves in the Body, the Safety and How Well Tolerated Different Increasing Amounts of the Drug ENTR-601-44 Are When Given to Healthy Male Volunteers; ISRCTN: London, UK, 2024; MAC Clinical Research; Available online: http://isrctn.com/ (accessed on 16 June 2025).
- Komaki, H.; Takeshita, E.; Kunitake, K.; Ishizuka, T.; Shimizu-Motohashi, Y.; Ishiyama, A.; Sasaki, M.; Yonee, C.; Maruama, S.; Hida, E.; et al. Phase 1/2 trial of brogidirsen: Dual-targeting antisense oligonucleotides for exon 44 skipping in Duchenne muscular dystrophy. Cell Rep. Med. 2025, 6, 101901. [Google Scholar] [CrossRef] [PubMed]
- Nippon Shinyaku Co., Ltd. FDA Grants Breakthrough Therapy Designation to NS-089/NCNP-02 for the Treatment of Duchenne Muscular Dystrophy. Nippon Shinyaku Co., Ltd. 28 July 2023. Available online: https://www.ncnp.go.jp/topics/2022/20220317e.html (accessed on 16 June 2025).
- Komaki, H.; Takeshita, E.; Kunitake, K.; Shimizu-Motohashi, Y.; Sasaki, M.; Yonee, C.; Maruyama, S.; Hida, E.; Matsubara, D.; Hatakeyama, T.; et al. P.123 A Phase I/II study of NS-089/NCNP-02, Exon 44 skipping drug, in patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2022, 32, S99–S100. [Google Scholar] [CrossRef]
- Shimo, T.; Maruyama, R.; Yokota, T. Designing Effective Antisense Oligonucleotides for Exon Skipping. In Duchenne Muscular Dystrophy: Methods and Protocols; Bernardini, C., Ed.; Humana Press: New York, NY, USA, 2018; Available online: http://www.springer.com/series/7651 (accessed on 16 June 2025).
- Watanabe, N.; Tone, Y.; Nagata, T.; Masuda, S.; Saito, T.; Motohashi, N.; Takagaki, K.; Aoki, Y.; Takeda, S. Exon 44 skipping in Duchenne muscular dystrophy: NS-089/NCNP-02, a dual-targeting antisense oligonucleotide. Mol. Ther. Nucleic Acids 2023, 34, 102034. [Google Scholar] [CrossRef]
- Komaki, H.; Takeshima, Y.; Matsumura, T.; Ozasa, S.; Funato, M.; Takeshita, E.; Iwata, Y.; Yajima, H.; Egawa, Y.; Toramoto, T.; et al. Viltolarsen in Japanese Duchenne muscular dystrophy patients: A phase 1/2 study. Ann. Clin. Transl. Neurol. 2020, 7, 2393–2408. [Google Scholar] [CrossRef]
- Ishizuka, T.; Komaki, H.; Asahina, Y.; Nakamura, H.; Motohashi, N.; Takeshita, E.; Shimizu-Motohashi, Y.; Ishiyama, A.; Tonee, C.; Maruyama, S.; et al. Systemic administration of the antisense oligonucleotide NS<-089/NCNP-02 for skipping of exon 44 in patients with Duchenne muscular dystrophy: Study protocol for a phase I/II clinical trial. Neuropsychopharmacol. Rep. 2023, 43, 277–286. [Google Scholar]
- Broomfield, J.; Abrams, K.; Latimer, N.; Guglieri, M.; Rutherford, M.; Crowther, M. Natural history of Duchenne muscular dystrophy in the United Kingdom: A descriptive study using the Clinical Practice Research Datalink. Brain Behav. 2023, 13, e3331. [Google Scholar] [CrossRef]
- Tulangekar, A.; Sztal, T.E. Inflammation in Duchenne Muscular Dystrophy–Exploring the Role of Neutrophils in Muscle Damage and Regeneration. Biomedicines 2021, 9, 1366. [Google Scholar] [CrossRef]
- Holland, A.; Lonkar, P.; Sweeney, C.; Zhang, H.; Svenstrup, N.; Gibbons, C.; Xu, L.; Joy, J.; Goyal, J. P27 Three novel enhanced delivery Oligonucleotide candidates for Duchenne muscular dystrophy mediate high levels of exon 53, 45, and 44 skipping. Neuromuscul. Disord. 2023, 33, S103–S104. [Google Scholar] [CrossRef]
- Mellion, M.; Larkindale, J.; Lonkar, P.; Goyal, J.; Holland, A.; Foy, J.; Garg, B.; Yu, S.; Frank, A.; Abbott, C.; et al. PGN-EDO51, an Enhanced Delivery Oligonucleotide (EDO) for the Treatment of Duchenne Muscular Dystrophy (DMD): Results of a Phase 1 Study in Healthy Volunteers. American Academy of Neurology 2023 Annual Meeting. American Academy of Neurology. 2023. Available online: https://www.aan.com/MSA/Public/Events/AbstractDetails/54382 (accessed on 25 June 2025).
- Shah, M.N.A.; Wilton-Clark, H.; Haque, F.; Powell, B.; Sutanto, L.E.; Maradiya, R.; Zhabyeyev, P.; Roshimi, R.R.; Anwar, S.; Aslesh, T.; et al. DG9 boosts PMO nuclear uptake and exon skipping to restore dystrophic muscle and cardiac function. Nat. Commun. 2025, 16, 4477. [Google Scholar] [CrossRef]
- Shang, M.; Wu, Y.; Wang, Y.; Cai, Y.; Jin, J.; Yang, Z. Dual antisense oligonucleotide targeting miR-21/miR-155 synergize photodynamic therapy to treat triple-negative breast cancer and inhibit metastasis. Biomed. Pharmacother. 2022, 146, 112564. [Google Scholar] [CrossRef]
- Moulton, H.M.; Moulton, J.D. Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim. et Biophys. Acta (BBA) Biomembr. 2010, 1798, 2296–2303. [Google Scholar] [CrossRef]
- Haque, U.S.; Yokota, T. Enhancing Antisense Oligonucleotide-Based Therapeutic Delivery with DG9, a Versatile Cell-Penetrating Peptide. Cells 2023, 12, 2395. [Google Scholar] [CrossRef]
- Waldrop, M.A.; Yaou, R.B.; Lucas, K.K.; Martin, A.S.; O’Rourke, E.; Filnemus; Ferlini, A.; Muntoni, F.; Leturcq, F.; Tuffery-Giraud, S.; et al. Clinical Phenotypes of DMD Exon 51 Skip Equivalent Deletions: A Systematic Review. J. Neuromuscul. Dis. 2020, 7, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Woo, S.; Melo, D.; Huang, Y.; Dzierlega, K.; Shah, M.N.A.; Aslesh, T.; Roshmi, R.R.; Echigoya, Y.; Maruyama, R.; et al. Development of DG9 peptide-conjugated single- and multi-exon skipping therapies for the treatment of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2022, 119, e2112546119. [Google Scholar] [CrossRef] [PubMed]
- Béroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizaed, M.P.; Voelckel, M.A.; Calemard, L.M.; Boisseau, P.; et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Van Deutekom, J.C.T.; Van Ommen, G.J.B. Advances in Duchenne muscular dystrophy gene therapy. Nat. Rev. Genet. 2003, 4, 774–783. [Google Scholar] [CrossRef]
- Anthony, K.; Cirak, S.; Torelli, S.; Tasca, G.; Feng, L.; Arechavala-Gomeza, V.; Armaroli, A.; Guglieri, M.; Straathof, C.S.; Verschuuren, J.J.; et al. Dystrophin quantification and clinical correlations in Becker muscular dystrophy: Implications for clinical trials. Brain 2011, 134, 3547–3559. [Google Scholar] [CrossRef]
- Heo, Y.-A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Relizani, K.; Griffith, G.; Echevarría, L.; Zarrouki, F.; Facchinetti, P.; Vaillend, C.; Leumann, C.; Garcia, L.; Goyenyalle, A. Efficacy and Safety Profile of Tricyclo-DNA Antisense Oligonucleotides in Duchenne Muscular Dystrophy Mouse Model. Mol. Ther. Nucleic Acids 2017, 8, 144–157. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Shieh, P.B.; Abdel-Hamid, H.Z.; Connolly, A.M.; Ciafaloni, E.; Wagner, K.R.; Goemans, N.; Mercuri, E.; Khan, N.; Koenig, E.; et al. Open-Label Evaluation of Eteplirsen in Patients with Duchenne Muscular Dystrophy Amenable to Exon 51 Skipping: PROMOVI Trial. J. Neuromuscul. Dis. 2021, 8, 989–1001. [Google Scholar] [CrossRef]
- Servais, L.; Mercuri, E.; Straub, V.; Guglieri, M.; Seferian, A.M.; Scoto, M.; Leone, D.; Koenig, E.; Khan, N.; Dugar, A.; et al. Long-Term Safety and Efficacy Data of Golodirsen in Ambulatory Patients with Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A First-in-human, Multicenter, Two-Part, Open-Label, Phase 1/2 Trial. Nucleic Acid Ther. 2022, 32, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.; Yokota, T. Duchenne muscular dystrophy: Promising early-stage clinical trials to watch. Expert Opin. Investig. Drugs 2024, 33, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Dovari, A.; Inuganti, B.; Nadimpally, J.; Vatturi, S.M.; Hyderboini, R.; Goyal, R. PRO38 Twenty Years of Clinical Trials in Duchenne Muscular Dystrophy: A Low Clinical Drug Development Success. Value Health 2021, 24, S204. [Google Scholar] [CrossRef]
- Lek, A.; Wong, B.; Keeler, A.; Blackwood, M.; Ma, K.; Huang, S.; Sylvia, K.; Batista, R.; Artinian, R.; Kokoski, D.; et al. Death after High-Dose rAAV9 Gene Therapy in a Patient with Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 2023, 389, 1203–1210. [Google Scholar] [CrossRef]
- Bönnemann, C.G.; Belluscio, B.A.; Braun, S.; Morris, C.; Singh, T.; Muntoni, F. Dystrophin Immunity after Gene Therapy for Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 2023, 388, 2294–2296. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| ClinicalTrials.gov ID | NCT04129294 | NCT05135663 | NCT05996003 |
|---|---|---|---|
| Phase | I/II | II | II |
| Start date | December 2019 | June 2021 | February 2024 |
| (Estimated) End date | May 2022 | July 2026 | November 2025 |
| Description | Dose-escalation, open-label | Open-label, extension study | Open-label, multi-center |
| Dose | 80 mg/kg, 40 mg/kg | ||
| Primary Endpoint | Safety, tolerability | Safety | Safety, pharmacokinetics |
| Secondary Endpoints | Pharmacokinetics, efficacy | Efficacy | Efficacy |
| Enrollment numbers | 6 | 6 | 20 (6 in Cohort 1, 14 in Cohort 2) |
| Route | Intravenous | Intravenous | Intravenous |
| Life expectancy | At least one year | Same participants as NCT04129294 | N/A |
| Age Range | 8–17 | 8–17 | 4–15 |
| Design | 24 weeks of treatment, 12-week follow-up period | Administer once weekly for 216 weeks | Once weekly for 4 weeks at 1 of 3 doses, 24 weeks at MTD 1 after |
| Corticosteroid use | None, or at least 6 months of stable use | Same participants as NCT04129294 | Stable dose for at least 3 months |
| Ambulation Requirements | Ambulant | Same participants as NCT04129294 | Able to walk independently without devices |
| Exclusion Criteria | No DNA polymorphisms that could compromise therapy and pre-mRNA binding, FVC 1 < 50% of predicted, EF 1 < 40%, FS 1 < 25% based on ECHO 1, current infections, cardiomyopathy, liver/renal disease, previous severe drug allergy, continuous use of artificial respirator, previous use of investigational therapies | Same participants as NCT04129294 | Body weight of <20 kg, cardiomyopathy symptoms, use of anabolic steroids, use of other investigational drugs in the past three months, surgery in last 3 months, taken gene therapy or another exon skipping drug |
| Other Criteria | Adequate intact muscles for biopsy, able to give written informed consent, QTc 1 < 450 ms (<480 ms for subjects with Bundle Branch Block) | Must have participated in NCT04129294 | Able to complete the TTSTAND 1 without assistance in <20 s, |
| Test Timing | At the end of the treatment period (24 weeks) | Up to Week 243 | Baseline, Week 13, Week 25 |
| Genotype | Out-of-frame deletion(s) amenable to exon 44 skipping | Out-of-frame deletion(s) amenable to exon 44 skipping | Amenable to exon 44 skipping |
| Reference | [66] | [72] | NCT05996003 |
| Exon-Skipping Therapies | AOC1044 (Delpacibart Zotadirsen) | ENTR-601-44 | NS-089/NCNP-02 (Brogidirsen) |
|---|---|---|---|
| Modification | Antibody-conjugated | Peptide-conjugated | Unmodified |
| Details | 4 PMOs conjugated to anti-TfR1 | PMO conjugated to EEV | No conjugations |
| Targeting Mechanism | TfR1 for targeted delivery | EEV to escape endosomes | Natural biodistribution |
| Target site(s) | Single | Single | Dual |
| Regulatory State | No clinical holds | FDA hold lifted in 2025 | No clinical holds |
| Current Clinical Stage | Phase II (Part B of EXPLORE44-OLE, enrolling DMD patients) | Phase Ib (ELEVATE-44-102 trial, enrolling DMD patients) | Phase II (NCT05996003, recruiting DMD patients) |
| Age | 7 to 27 years old | ||
| Reported clinical trial design(s) | 40 healthy volunteers in a double-blind, placebo-controlled trial | 4 dose levels tested in healthy males | 4 dose levels tested in 6 DMD patients |
| 25 patients receiving AOC1044 | 32 patients to be enrolled | 20 patients to be enrolled |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, A.; Yokota, T. Brogidirsen and Exon 44 Skipping for Duchenne Muscular Dystrophy: Advances and Challenges in RNA-Based Therapy. Genes 2025, 16, 777. https://doi.org/10.3390/genes16070777
Tang A, Yokota T. Brogidirsen and Exon 44 Skipping for Duchenne Muscular Dystrophy: Advances and Challenges in RNA-Based Therapy. Genes. 2025; 16(7):777. https://doi.org/10.3390/genes16070777
Chicago/Turabian StyleTang, Annie, and Toshifumi Yokota. 2025. "Brogidirsen and Exon 44 Skipping for Duchenne Muscular Dystrophy: Advances and Challenges in RNA-Based Therapy" Genes 16, no. 7: 777. https://doi.org/10.3390/genes16070777
APA StyleTang, A., & Yokota, T. (2025). Brogidirsen and Exon 44 Skipping for Duchenne Muscular Dystrophy: Advances and Challenges in RNA-Based Therapy. Genes, 16(7), 777. https://doi.org/10.3390/genes16070777