
Taxonomic Diversity and Clinical Correlations in Periapical Lesions by Next-Generation Sequencing Analysis


Abstract
1. Introduction
2. Methods
2.1. Search for Available Sequences in Databases
2.2. DADA2 Read Processing
2.3. Taxonomy Assignment
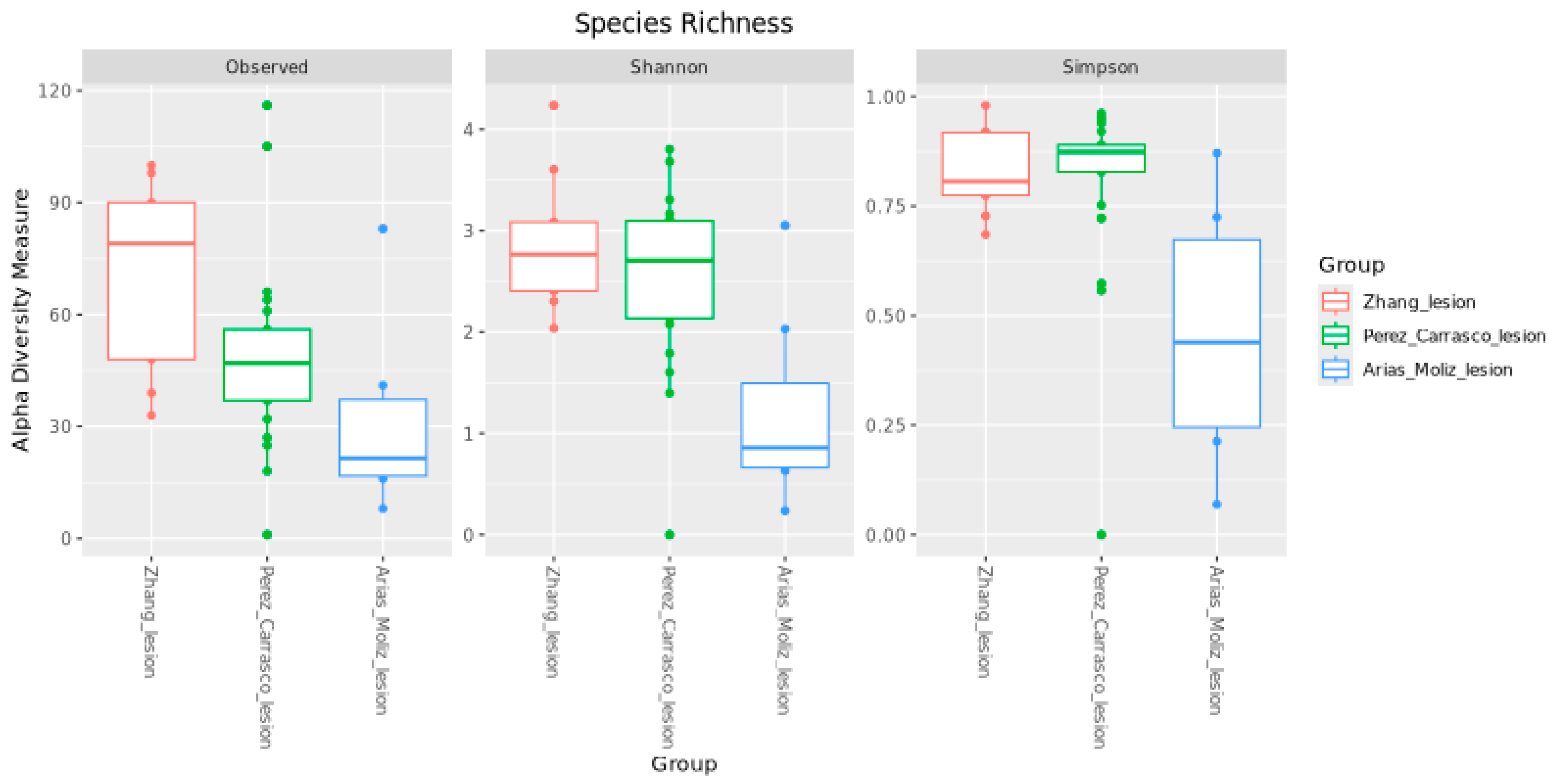
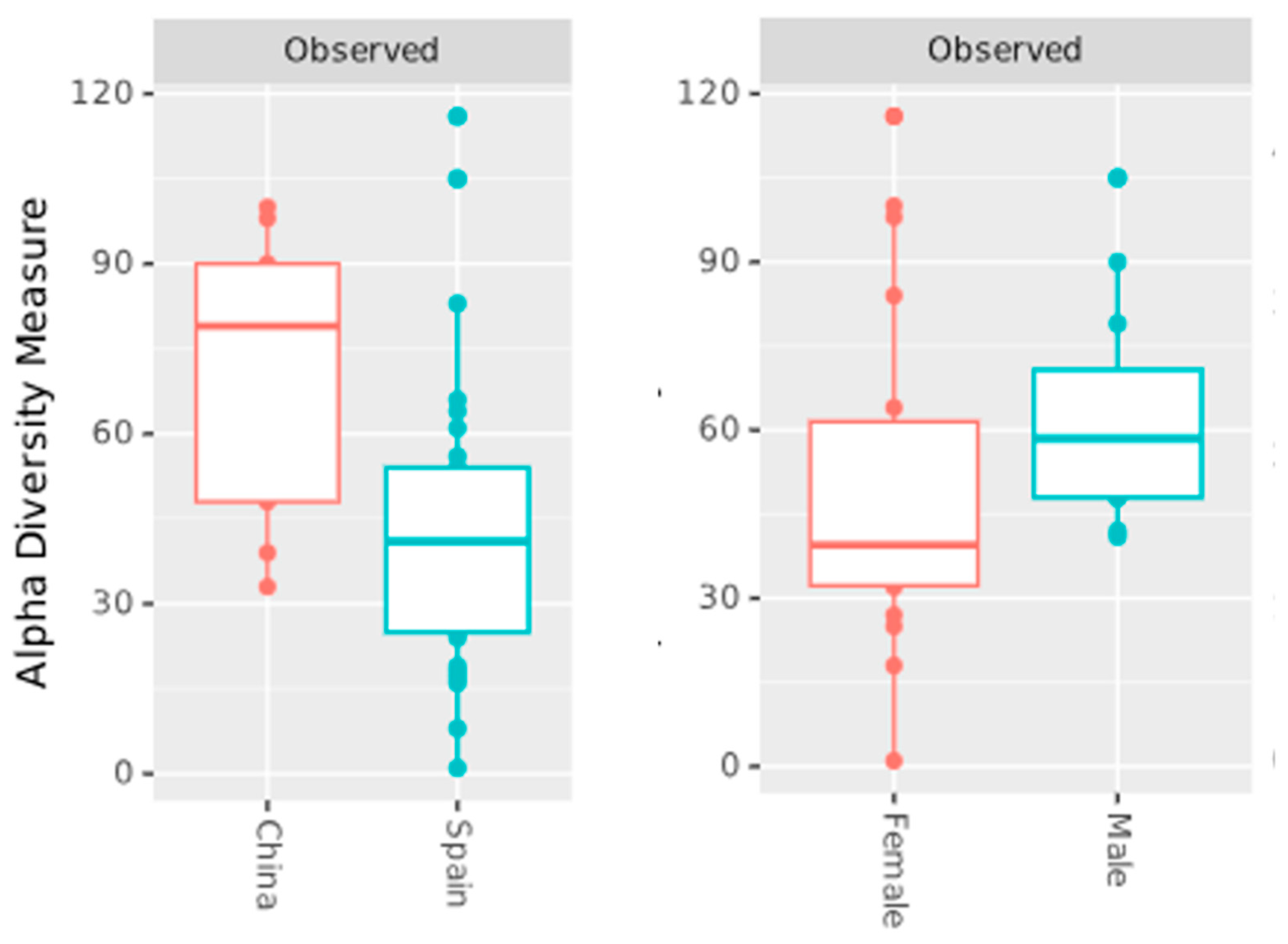
2.4. Alpha Diversity
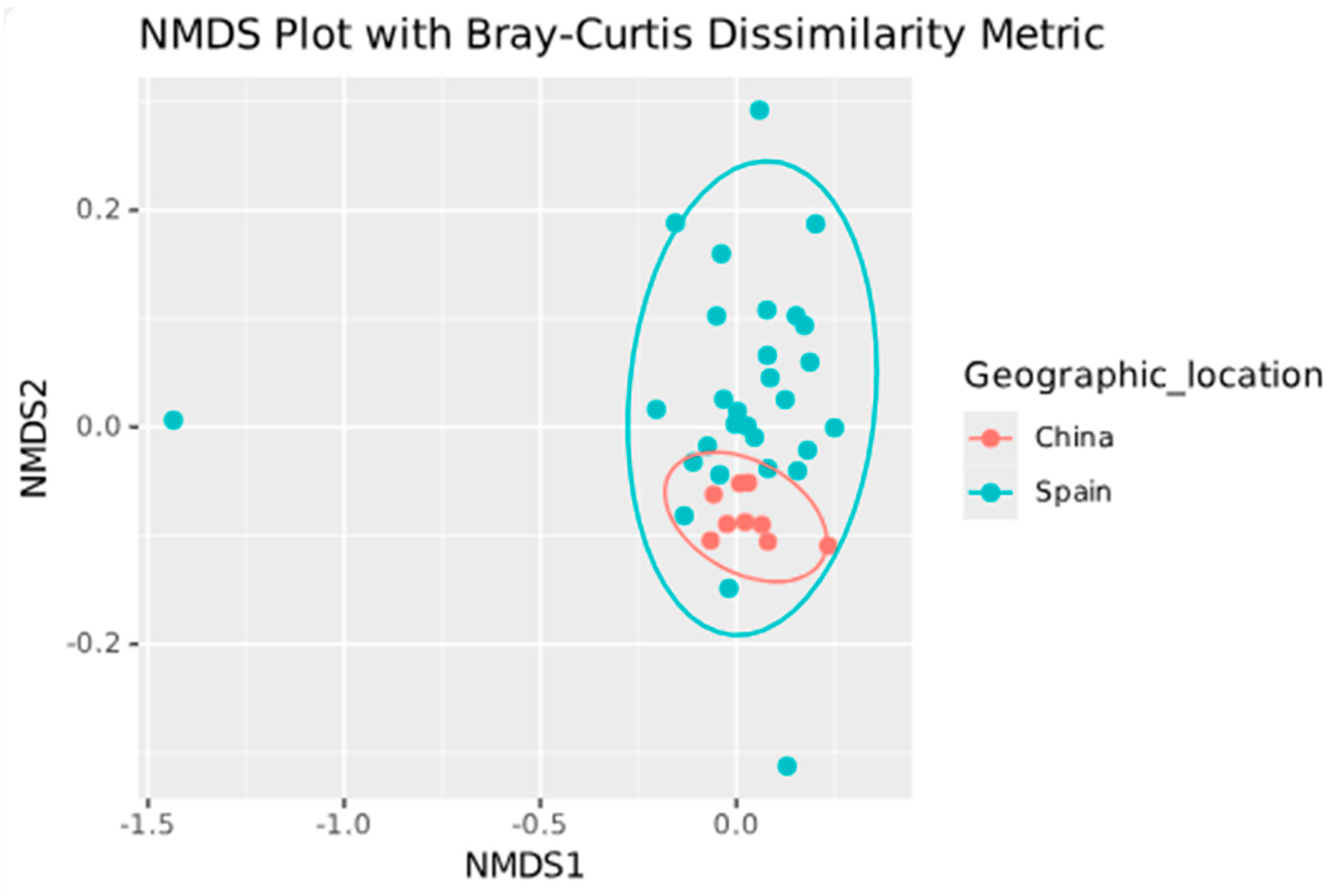
2.5. Beta Diversity
2.6. Differential Abundance (ANCOM-BC2)
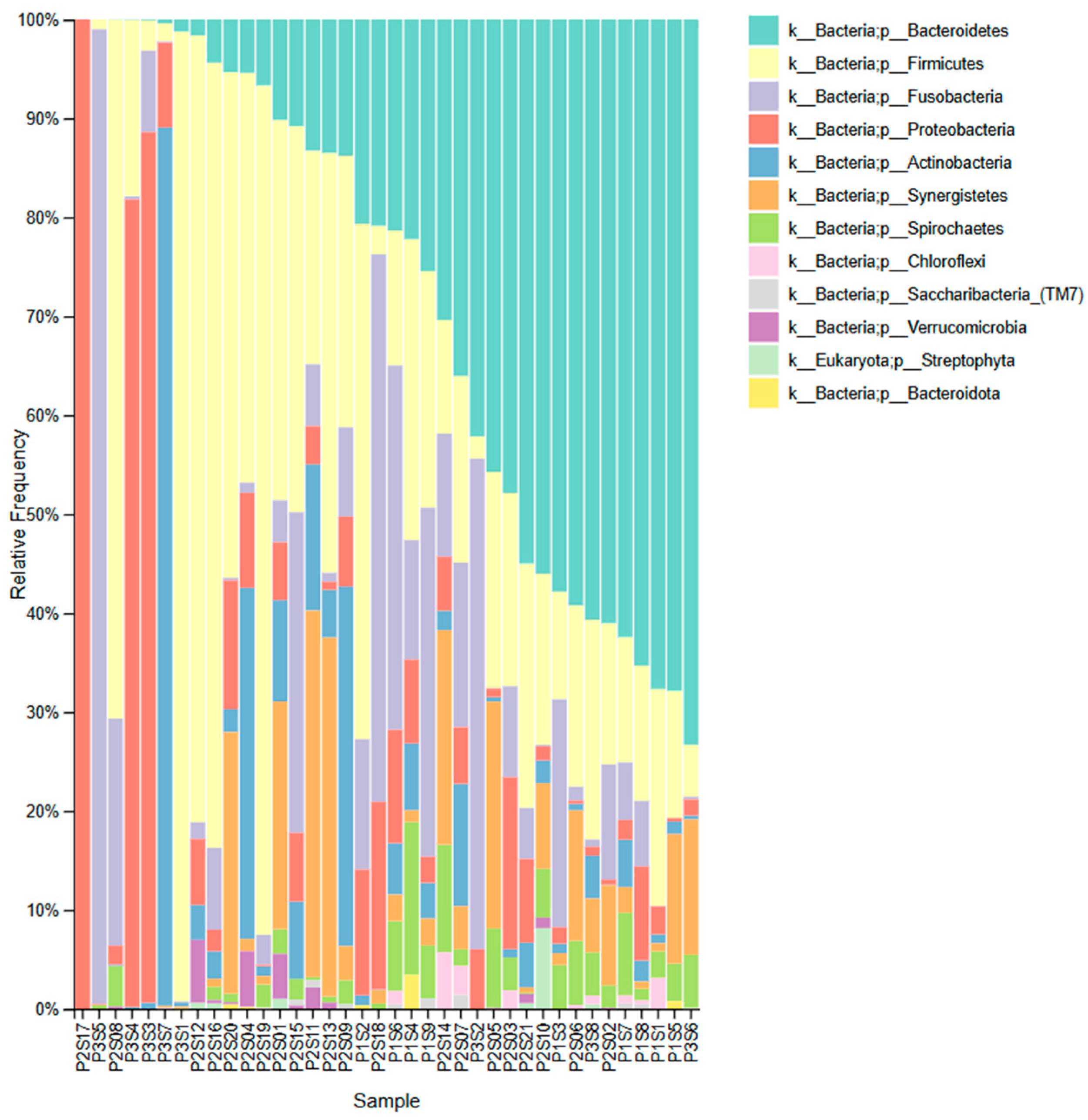
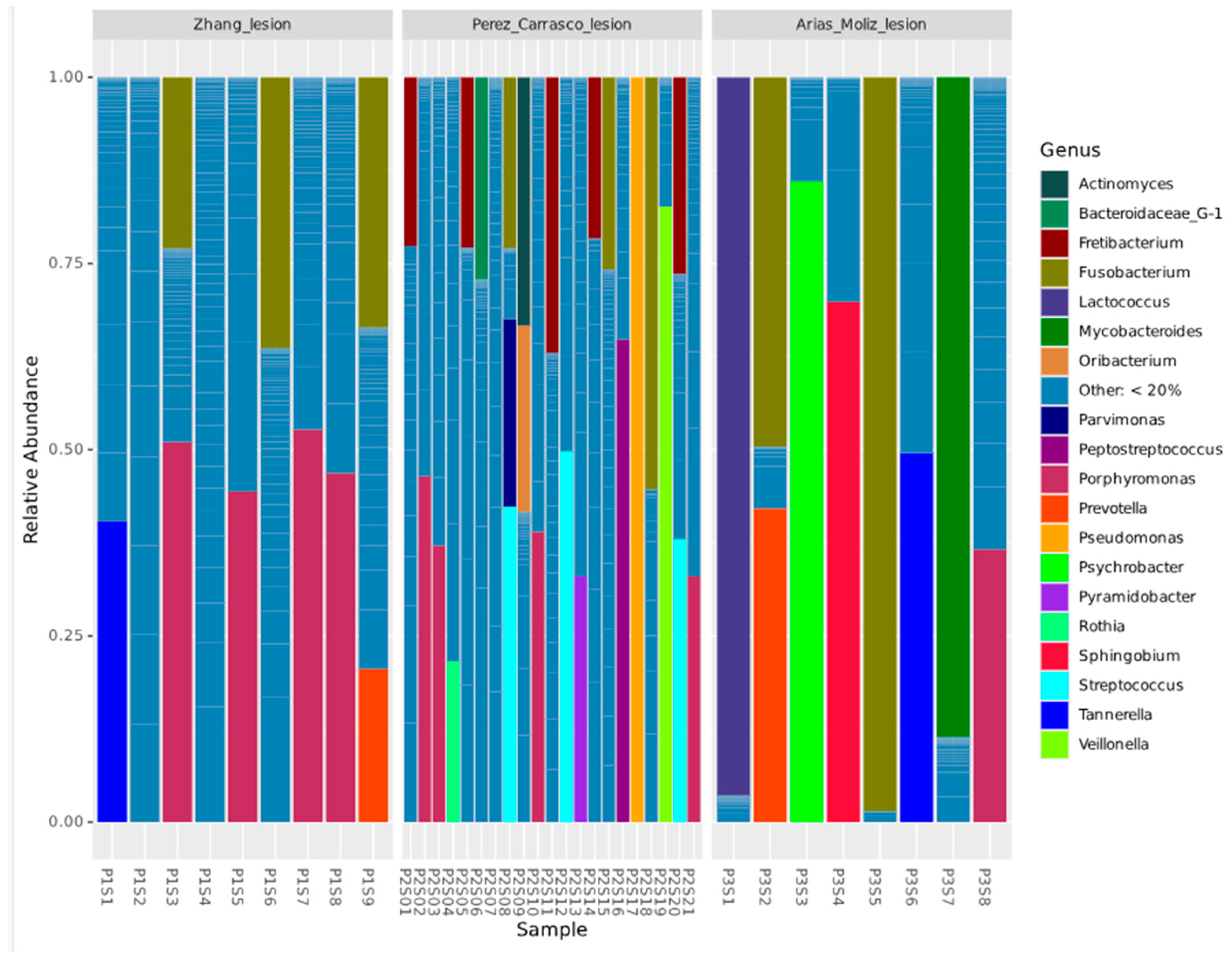
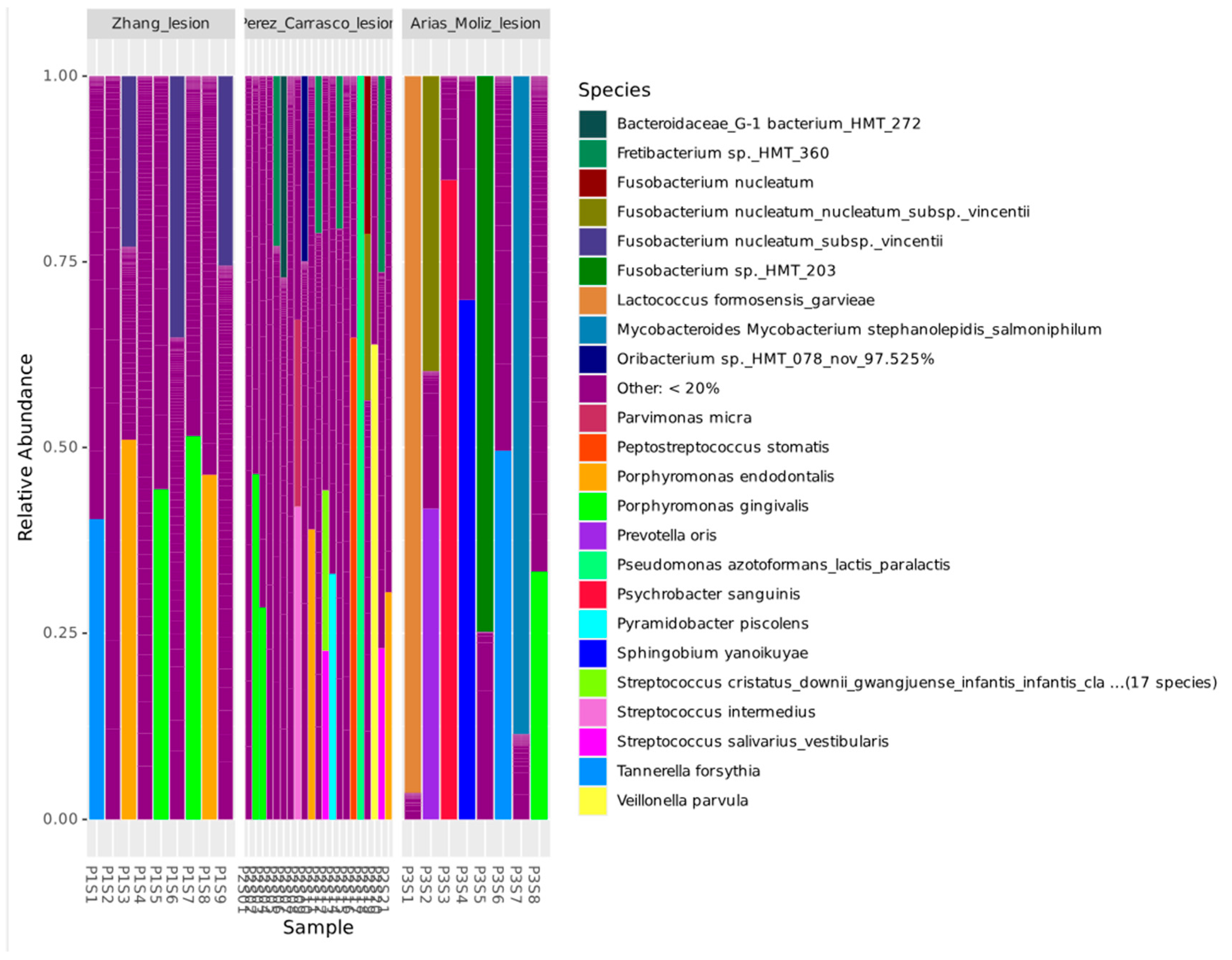
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kakehashi, S.; Stanley, H.R.; Fitzgerald, R.J. The effects of surgical exposures of dental pulps in germ-free and conventional laboratory rats. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 1965, 20, 340–349. [Google Scholar] [CrossRef] [PubMed]
- American Association of Endodontists. Glossary of Endodontic Terms, 10th ed.; American Association of Endodontists: Chicago, IL, USA, 2020; p. 37. [Google Scholar]
- Estrela, C.; Holland, R.; Estrela, C.R.; Alencar, A.H.; Sousa-Neto, M.D.; Pecora, J.D. Characterization of successful root canal treatment. Braz. Dent. J. 2014, 25, 3–11. [Google Scholar] [CrossRef]
- Tibúrcio-Machado, C.S.; Michelon, C.; Zanatta, F.B.; Gomes, M.S.; Marin, J.A.; Bier, C.A. The global prevalence of apical periodontitis: A systematic review and meta-analysis. Int. Endod. J. 2021, 54, 712–735. [Google Scholar] [CrossRef] [PubMed]
- Bronzato, J.D.; Bomfim, R.A.; Hayasida, G.Z.P.; Cúri, M.; Estrela, C.; Paster, B.J.; Gomes, B.P.F.A. Analysis of microorganisms in periapical lesions: A systematic review and meta-analysis. Arch. Oral. Biol. 2021, 124, 105055. [Google Scholar] [CrossRef]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef]
- Han, H.; Lee, H.J.; Kim, K.S.; Chung, J.; Na, H.S. Comparison of the performance of MiSeq and NovaSeq in oral microbiome study. J. Oral. Microbiol. 2024, 16, 2344293. [Google Scholar] [CrossRef]
- Siqueira, J.F., Jr.; Rôças, I.N. A critical analysis of research methods and experimental models to study the root canal microbiome. Int. Endod. J. 2022, 55 (Suppl. 1), 46–71. [Google Scholar] [CrossRef]
- Arias-Moliz, M.T.; Pérez-Carrasco, V.; Uroz-Torres, D.; Santana Ramos, J.D.; García-Salcedo, J.A.; Soriano, M. Identification of keystone taxa in root canals and periapical lesions of post-treatment endodontic infections: Next generation microbiome research. Int. Endod. J. 2024, 57, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Carrasco, V.; Uroz-Torres, D.; Soriano, M.; Solana, C.; Ruiz-Linares, M.; Garcia-Salcedo, J.A.; Arias-Moliz, M.T. Microbiome in paired root apices and periapical lesions and its association with clinical signs in persistent apical periodontitis using next-generation sequencing. Int. Endod. J. 2023, 56, 622–636. [Google Scholar] [CrossRef]
- Zhang, J.L.; Yun, J.; Yue, L.; Du, W.; Liang, Y.H. Distinctive microbiota distribution from healthy oral to post-treatment apical periodontitis. Front. Cell Infect. Microbiol. 2022, 12, 980157. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Chen, T.; Yu, W.H.; Izard, J.; Baranova, O.V.; Lakshmanan, A.; Dewhirst, F.E. The Human Oral Microbiome Database: A web accessible resource for investigating oral microbe taxonomic and genomic information. Database 2010, 2010, baq013. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Al-Hebshi, N.N.; Nasher, A.T.; Idris, A.M.; Chen, T. Robust species taxonomy assignment algorithm for 16S rRNA NGS reads: Application to oral carcinoma samples. J. Oral. Microbiol. 2015, 7, 28934. [Google Scholar] [CrossRef]
- Whittaker, R.H. Evolution and measurement of species diversity. Taxon 1972, 21, 213–251. [Google Scholar] [CrossRef]
- Lin, H.; Peddada, S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef]
- Park, D.H.; Park, O.J.; Yoo, Y.J.; Perinpanayagam, H.; Cho, E.B.; Kim, K.; Park, J.; Noblett, W.C.; Kum, K.Y.; Han, S.H. Microbiota association and profiling of gingival sulci and root canals of teeth with primary or secondary/persistent endodontic infections. J. Endod. 2024, 50, 1124–1133. [Google Scholar] [CrossRef]
- Bronzato, J.D.; Davidian, M.E.S.; de Castro, M.; de-Jesus-Soares, A.; Ferraz, C.C.R.; Almeida, J.F.A.; Marciano, M.A.; Gomes, B.P.F.A. Bacteria and virulence factors in periapical lesions associated with teeth following primary and secondary root canal treatment. Int. Endod. J. 2021, 54, 660–671. [Google Scholar] [CrossRef]
- Ordinola-Zapata, R.; Costalonga, M.; Nixdorf, D.; Dietz, M.; Schuweiler, D.; Lima, B.P.; Staley, C. Taxonomic abundance in primary and secondary root canal infections. Int. Endod. J. 2023, 56, 278–288. [Google Scholar] [CrossRef]
- Hong, L.; Hai, J.; Yan-Yan, H.; Shenghui, Y.; Benxiang, H. Colonization of Porphyromonas endodontalis in primary and secondary endodontic infections. Hua Xi Kou Qiang Yi Xue Za Zhi = Huaxi Kouqiang Yixue Zazhi = West China J. Stomatol. 2015, 33, 88–92. [Google Scholar] [CrossRef]
- Alquria, T.A.; Acharya, A.; Kabir, B.; Griffin, I.L.; Tordik, P.A.; Martinho, F.C. Clinical investigation of bacteriome in primary endodontic infections with apical periodontitis using high-throughput sequencing analysis. J. Endod. 2024, 50, 1393–1402. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef]
- Darveau, R.P.; Hajishengallis, G.; Curtis, M.A. Porphyromonas gingivalis as a potential community activist for disease. J. Dent. Res. 2012, 91, 816–820. [Google Scholar] [CrossRef]
- Kolenbrander, P.E.; Palmer, R.J., Jr.; Periasamy, S.; Jakubovics, N.S. Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 2010, 8, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Bronzato, J.D.; Bomfim, R.A.; Edwards, D.H.; Crouch, D.; Hector, M.P.; Gomes, B.P.F.A. Detection of Fusobacterium in oral and head and neck cancer samples: A systematic review and meta-analysis. Arch. Oral. Biol. 2020, 112, 104669. [Google Scholar] [CrossRef]
- Gomes, B.P.F.A.; Bronzato, J.D.; Almeida-Gomes, R.F.; Pinheiro, E.T.; Sousa, E.L.R.; Jacinto, R.C. Identification of Fusobacterium nucleatum in primary and secondary endodontic infections and its association with clinical features by using two different methods. Clin. Oral. Investig. 2021, 25, 6249–6258. [Google Scholar] [CrossRef]
- Emerson, J.B.; Adams, R.I.; Roman, C.M.B.; Brooks, B.; Coil, D.A.; Dahlhausen, K.; Ganz, H.H.; Hartmann, E.M.; Hsu, T.; Justice, N.B.; et al. Schrodinger’s microbes: Tools for distinguishing the living from the dead in microbial ecosystems. Microbiome 2017, 5, 86. [Google Scholar] [CrossRef]
- Sunde, P.T.; Olsen, I.; Göbel, U.B.; Theegarten, D.; Winter, S.; Debelian, G.J.; Tronstad, L.; Moter, A. Fluorescence in situ hybridization (FISH) for direct visualization of bacteria in periapical lesions of asymptomatic root-filled teeth. Microbiology 2003, 149, 1095–1102. [Google Scholar] [CrossRef]
- Nakano, M. 16S rRNA gene primer validation for bacterial diversity analysis of vegetable products. J. Food Prot. 2018, 81, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Erb-Downward, J.R.; Falkowski, N.R.; D’Souza, J.C.; McCloskey, L.M.; McDonald, R.A.; Brown, C.A.; Shedden, K.; Dickson, R.P.; Freeman, C.M.; Stringer, K.A.; et al. Critical relevance of stochastic effects on low-bacterial-biomass 16S rRNA gene analysis. mBio 2020, 11, e00598-21. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, E.T.; Karygianni, L.; Candeiro, G.T.M.; Vilela, B.G.; Dantas, L.O.; Pereira, A.C.C.; Gomes, B.; Attin, T.; Thurnheer, T.; Russo, G. Metatranscriptome and resistome of the endodontic microbiome. J. Endod. 2024, 50, 1059–1072.e1054. [Google Scholar] [CrossRef] [PubMed]
- Ordinola-Zapata, R.; Costalonga, M.; Dietz, M.; Lima, B.P.; Staley, C. The root canal microbiome diversity and function. A whole-metagenome shotgun analysis. Int. Endod. J. 2024, 57, 872–884. [Google Scholar] [CrossRef]
- Nardello, L.C.L.; Amado, P.P.P.; Franco, D.C.; Cazares, R.X.R.; Nogales, C.G.; Mayer, M.P.A.; Karygianni, L.; Thurnheer, T.; Pinheiro, E.T. Next-generation sequencing to assess potentially active bacteria in endodontic infections. J. Endod. 2020, 46, 1105–1112. [Google Scholar] [CrossRef]
- Sharma, G.; Garg, N.; Hasan, S.; Shirodkar, S. Prevotella: An insight into its characteristics and associated virulence factors. Microb. Pathog. 2022, 169, 105673. [Google Scholar] [CrossRef]
- Könönen, E.; Fteita, D.; Gursoy, U.K.; Gursoy, M. Prevotella species as oral residents and infectious agents with potential impact on systemic conditions. J. Oral. Microbiol. 2022, 14, 2079814. [Google Scholar] [CrossRef]
- Arias-Moliz, M.T.; Ordinola-Zapata, R.; Staley, C.; Pérez-Carrasco, V.; García-Salcedo, J.A.; Uroz-Torres, D.; Soriano, M. Exploring the root canal microbiome in previously treated teeth: A comparative study of diversity and metabolic pathways across two geographical locations. Int. Endod. J. 2024, 57, 885–894. [Google Scholar] [CrossRef]
- Yan, Z.; Zheng, Z.; Xia, T.; Ni, Z.; Dou, Y.; Liu, X. Causal relationship between gut microbiome and sex hormone-binding globulin: A bidirectional two-sample Mendelian randomization study. Am. J. Reprod. Immunol. 2024, 91, e13824. [Google Scholar] [CrossRef]
- Sunde, P.T.; Olsen, I.; Lind, P.O.; Tronstad, L. Extraradicular infection: A methodological study. Endod. Dent. Traumatol. 2000, 16, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Singer, G.A.C.; Fahner, N.A.; Barnes, J.G.; McCarthy, A.; Hajibabaei, M. Comprehensive biodiversity analysis via ultra-deep patterned flow cell technology: A case study of eDNA metabarcoding seawater. Sci. Rep. 2019, 9, 5991. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Characteristics | Zhang et al. [11] | Perez-Carrasco et al. [10] | Arias-Moliz et al. [9] |
|---|---|---|---|
| Year | 2022 | 2023 | 2024 |
| Country | China | Spain | Spain |
| Journal | Frontiers in Cellular and Infection Microbiology | International Endodontic Journal | International Endodontic Journal |
| Study type | Observational | Observational | Observational |
| Bioproject registration | PRJNA808987 | PRJNA839210 | PRJNA1033443 |
| Industrial funding | No | No | No |
| Number of samples analyzed in the present study | 9 periapical lesions | 21 periapical lesions | 8 periapical lesions |
| Mean age (SD) | 36 (13.37) | 45.9 (13.9) | Unknown |
| Teeth type | Single and multi-rooted | Single, bi-, and multi-rooted | Unknown |
| Teeth localization | Upper and lower | Upper and lower | Unknown |
| Type of operator | Experienced endodontist | Experienced endodontist | Experienced endodontist |
| Number of previous root canal treatments | 1 to 2 | 1 | Unknown |
| Lesion collection | curette | curette and tweezers | curette and tweezers |
| Use of magnification | yes | yes | yes |
| Sample storage | Sterile PBS | Cryopulverization | Cryopulverization |
| Inclusion criteria | Endodontically treated teeth with adequate coronal restoration and true periapical lesions without involving the periodontal tissues (probing depth [PD] ≤3 mm). | Previously treated root canal for at least 1 year with radiographic evidence of persistent apical periodontitis. | Patients showing radiographic evidence of persistent apical periodontitis. |
| Exclusion criteria | History of antibiotic use in the past month, vertical root fracture, and resurgery teeth. | Severe systemic disease, use of antibiotics in the 3 months previous to surgery, teeth with periodontal pockets >4 mm, pulp cavity exposure to the oral environment, teeth with vertical root fracture, and history of trauma. Pregnant or breastfeeding women and patients under 18 years old were also excluded. | Patients with severe systemic diseases, pregnant or breastfeeding women, and patients under 18 years old. Teeth with periodontal pockets >4 mm, vertical root fracture, and history of trauma. |
| Illumina Sequencer | NovaSeq 6000 | MiSeq | MiSeq |
| Targeted 16S rRNA gene region | V3–V4 | V3–V4 | V3–V4 |
| Step | Zhang et al. [11]—Reads | Zhang et al.—% | Perez-Carrasco et al. [10]—Reads | Perez-Carrasco et al.—% | Arias-Moliz et al. [9]—Reads | Arias-Moliz et al.—% |
|---|---|---|---|---|---|---|
| Input | 771,878 | 100.0 | 353,875 | 100.0 | 198,448 | 100.0 |
| Filtered | 757,227 | 98.1 | 348,361 | 98.4 | 197,833 | 99.7 |
| DenoisedF | 735,235 | 95.3 | 324,606 | 91.7 | 152,593 | 76.9 |
| DenoisedR | 724,395 | 93.8 | 317,580 | 89.7 | 142,944 | 72.0 |
| Merged | 702,680 | 91.0 | 297,976 | 84.2 | 122,709 | 61.8 |
| Nonchim | 256,657 | 33.3 | 293,018 | 82.8 | 120,710 | 60.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bronzato, J.D.; Gomes, B.P.F.A.; Chen, T. Taxonomic Diversity and Clinical Correlations in Periapical Lesions by Next-Generation Sequencing Analysis. Genes 2025, 16, 775. https://doi.org/10.3390/genes16070775
Bronzato JD, Gomes BPFA, Chen T. Taxonomic Diversity and Clinical Correlations in Periapical Lesions by Next-Generation Sequencing Analysis. Genes. 2025; 16(7):775. https://doi.org/10.3390/genes16070775
Chicago/Turabian StyleBronzato, Juliana D., Brenda P. F. A. Gomes, and Tsute Chen. 2025. "Taxonomic Diversity and Clinical Correlations in Periapical Lesions by Next-Generation Sequencing Analysis" Genes 16, no. 7: 775. https://doi.org/10.3390/genes16070775
APA StyleBronzato, J. D., Gomes, B. P. F. A., & Chen, T. (2025). Taxonomic Diversity and Clinical Correlations in Periapical Lesions by Next-Generation Sequencing Analysis. Genes, 16(7), 775. https://doi.org/10.3390/genes16070775