
Phenotypic and Genotypic Characterization of 171 Patients with Syndromic Inherited Retinal Diseases Highlights the Importance of Genetic Testing for Accurate Clinical Diagnosis

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Clinical Evaluation
2.3. Genetic Analyses
2.4. Bioinformatics
3. Results
3.1. Characteristics of the Syndromic IRD Cohort
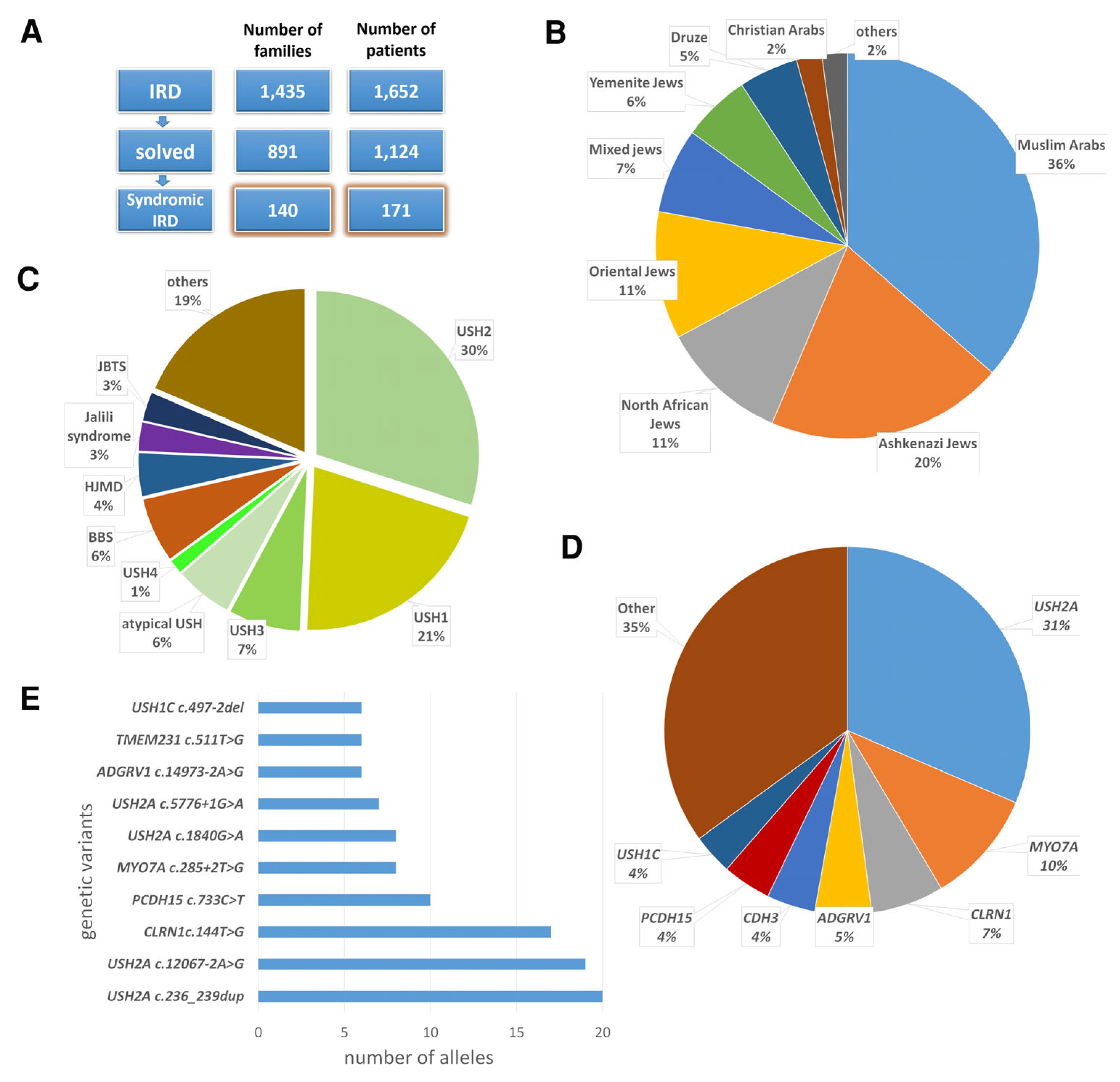
3.2. Revised Clinical Diagnosis Following Genetic Analysis
3.3. Co-Occurrence of Syndromic IRD and Additional Phenotypes
3.4. New Genotype–Phenotype Correlation: Biallelic Pathogenic Variants in KATNIP in an Individual with a JBTS26-Related Ciliopathy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Autosomal dominant |
| AF | Allele frequency |
| AR | Autosomal recessive |
| BBS | Bardet–Biedl syndrome |
| BCVA | Best-corrected visual acuity |
| CRD | Cone–rod dystrophy |
| DD | Developmental delay |
| FAF | Fundus autofluorescence |
| ffERG | Full-field electroretinogram |
| HL | Hearing loss |
| HMLR2 | Heimler syndrome 2 |
| ID | Intellectual disability |
| IEM | Inborn errors of metabolism |
| IRB | Institutional review board |
| IRD | Inherited retinal disease |
| JBTS | Joubert syndrome |
| LCAEOD | Leber congenital amaurosis with early-onset deafness |
| mfERG | Multi-focal electroretinogram |
| OCT | Optical coherence tomography |
| PBD4A | Peroxisomal biogenesis disorder 4A |
| PCD | Primary ciliary dyskinesia |
| RP | Retinitis pigmentosa |
| SNHL | Sensorineural hearing loss |
| USH | Usher syndrome |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
| y | Years |
References
- Ben-Yosef, T. Inherited Retinal Diseases. Int. J. Mol. Sci. 2022, 23, 13467. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Sundaresan, Y.; Gopalakrishnan, P.; Beryozkin, A.; Hanany, M.; Levanon, E.Y.; Banin, E.; Ben-Aroya, S.; Sharon, D. Inherited retinal diseases: Linking genes, disease-causing variants, and relevant therapeutic modalities. Prog. Retin. Eye Res. 2021, 89, 101029. [Google Scholar] [CrossRef] [PubMed]
- Tatour, Y.; Ben-Yosef, T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics 2020, 10, 779. [Google Scholar] [CrossRef] [PubMed]
- Delmaghani, S.; El-Amraoui, A. The genetic and phenotypic landscapes of Usher syndrome: From disease mechanisms to a new classification. Hum. Genet. 2022, 141, 709–735. [Google Scholar] [CrossRef]
- Sreekumar, V.; Norris, D.P. Cilia and development. Curr. Opin. Genet. Dev. 2019, 56, 15–21. [Google Scholar] [CrossRef]
- Forsyth, R.; Gunay-Aygun, M. Bardet-Biedl Syndrome Overview. In GeneReviews((R)); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2003. [Google Scholar]
- Forsythe, E.; Beales, P.L. Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef]
- Glass, I.A.; Dempsey, J.C.; Parisi, M.; Doherty, D. Joubert Syndrome. In GeneReviews((R)); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Parisi, M.A.; Doherty, D.; Chance, P.F.; Glass, I.A. Joubert syndrome (and related disorders) (OMIM 213300). Eur. J. Hum. Genet. 2007, 15, 511–521. [Google Scholar] [CrossRef]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Senior-Loken Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 175–178. [Google Scholar] [CrossRef]
- Luscan, R.; Mechaussier, S.; Paul, A.; Tian, G.; Gerard, X.; Defoort-Dellhemmes, S.; Loundon, N.; Audo, I.; Bonnin, S.; LeGargasson, J.F.; et al. Mutations in TUBB4B Cause a Distinctive Sensorineural Disease. Am. J. Hum. Genet. 2017, 101, 1006–1012. [Google Scholar] [CrossRef]
- Dodd, D.O.; Mechaussier, S.; Yeyati, P.L.; McPhie, F.; Anderson, J.R.; Khoo, C.J.; Shoemark, A.; Gupta, D.K.; Attard, T.; Zariwala, M.A.; et al. Ciliopathy patient variants reveal organelle-specific functions for TUBB4B in axonemal microtubules. Science 2024, 384, eadf5489. [Google Scholar] [CrossRef]
- Ferreira, C.R.; van Karnebeek, C.D.M. Inborn errors of metabolism. Handb. Clin. Neurol. 2019, 162, 449–481. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.J.; Dodt, G.; Raymond, G.V.; Braverman, N.E.; Moser, A.B.; Moser, H.W. Peroxisome biogenesis disorders. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2006, 1763, 1733–1748. [Google Scholar] [CrossRef] [PubMed]
- Ratbi, I.; Falkenberg, K.D.; Sommen, M.; Al-Sheqaih, N.; Guaoua, S.; Vandeweyer, G.; Urquhart, J.E.; Chandler, K.E.; Williams, S.G.; Roberts, N.A.; et al. Heimler Syndrome Is Caused by Hypomorphic Mutations in the Peroxisome-Biogenesis Genes PEX1 and PEX6. Am. J. Hum. Genet. 2015, 97, 535–545. [Google Scholar] [CrossRef]
- Yahraus, T.; Braverman, N.; Dodt, G.; Kalish, J.E.; Morrell, J.C.; Moser, H.W.; Valle, D.; Gould, S.J. The peroxisome biogenesis disorder group 4 gene, PXAAA1, encodes a cytoplasmic ATPase required for stability of the PTS1 receptor. EMBO J. 1996, 15, 2914–2923. [Google Scholar] [CrossRef]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations. Genes 2017, 8, 304. [Google Scholar] [CrossRef]
- Karali, M.; Testa, F.; Di Iorio, V.; Torella, A.; Zeuli, R.; Scarpato, M.; Romano, F.; Onore, M.E.; Pizzo, M.; Melillo, P.; et al. Genetic epidemiology of inherited retinal diseases in a large patient cohort followed at a single center in Italy. Sci. Rep. 2022, 12, 20815. [Google Scholar] [CrossRef]
- Marta, A.; Marques-Couto, P.; Vaz-Pereira, S.; Costa, J.; Cabral, D.; Estrela-Silva, S.; Franca, M.; Marques, J.H.; Meneres, M.J.; Lemos, C.; et al. Clinical and genetic landscape of IRD in Portugal: Pooled data from the nationwide IRD-PT registry. NPJ Genom. Med. 2025, 10, 11. [Google Scholar] [CrossRef]
- Perea-Romero, I.; Gordo, G.; Iancu, I.F.; Del Pozo-Valero, M.; Almoguera, B.; Blanco-Kelly, F.; Carreno, E.; Jimenez-Rolando, B.; Lopez-Rodriguez, R.; Lorda-Sanchez, I.; et al. Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci. Rep. 2021, 11, 1526. [Google Scholar] [CrossRef]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Gnanasekaran, H.; Chandrasekhar, S.P.; Kandeeban, S.; Periyasamy, P.; Bhende, M.; Khetan, V.; Gupta, N.; Kabra, M.; Namboothri, S.; Sen, P.; et al. Mutation profile of Bardet-Biedl syndrome patients from India: Implicative role of multiallelic rare variants and oligogenic inheritance pattern. Clin. Genet. 2023, 104, 443–460. [Google Scholar] [CrossRef] [PubMed]
- Kiraz, A.; Erdogan, M.; Balta, B.; Gumus, H.; Mutlu, M.B.; Mammadova, N.; Ozcelik, F.; Sahin, I.O.; Guven, A.S.; Kumandas, S.; et al. A case series of joubert syndrome evaluated with whole exome sequencing and the utility of optical genome mapping in the diagnosis. Neurogenetics 2025, 26, 47. [Google Scholar] [CrossRef] [PubMed]
- Perea-Romero, I.; Blanco-Kelly, F.; Sanchez-Navarro, I.; Lorda-Sanchez, I.; Tahsin-Swafiri, S.; Avila-Fernandez, A.; Martin-Merida, I.; Trujillo-Tiebas, M.J.; Lopez-Rodriguez, R.; Rodriguez de Alba, M.; et al. NGS and phenotypic ontology-based approaches increase the diagnostic yield in syndromic retinal diseases. Hum. Genet. 2021, 140, 1665–1678. [Google Scholar] [CrossRef]
- Auslender, N.; Bandah, D.; Rizel, L.; Behar, D.M.; Shohat, M.; Banin, E.; Allon-Shalev, S.; Sharony, R.; Sharon, D.; Ben-Yosef, T. Four USH2A founder mutations underlie the majority of Usher syndrome type 2 cases among non-Ashkenazi Jews. Genet. Test. 2008, 12, 289–294. [Google Scholar] [CrossRef]
- Ben-Yosef, T.; Asia Batsir, N.; Ali Nasser, T.; Ehrenberg, M. Retinal dystrophy as part of TTC21B-associated ciliopathy. Ophthalmic Genet. 2021, 42, 329–333. [Google Scholar] [CrossRef]
- Fadaie, Z.; Whelan, L.; Ben-Yosef, T.; Dockery, A.; Corradi, Z.; Gilissen, C.; Haer-Wigman, L.; Corominas, J.; Astuti, G.D.N.; de Rooij, L.; et al. Whole genome sequencing and in vitro splice assays reveal genetic causes for inherited retinal diseases. NPJ Genom. Med. 2021, 6, 97. [Google Scholar] [CrossRef]
- Goldenberg-Cohen, N.; Banin, E.; Zalzstein, Y.; Cohen, B.; Rotenstreich, Y.; Rizel, L.; Basel-Vanagaite, L.; Ben-Yosef, T. Genetic heterogeneity and consanguinity lead to a “double hit”: Homozygous mutations of MYO7A and PDE6B in a patient with retinitis pigmentosa. Mol. Vis. 2013, 19, 1565–1571. [Google Scholar]
- Khalaileh, A.; Abu-Diab, A.; Ben-Yosef, T.; Raas-Rothschild, A.; Lerer, I.; Alswaiti, Y.; Chowers, I.; Banin, E.; Sharon, D.; Khateb, S. The Genetics of Usher Syndrome in the Israeli and Palestinian Populations. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1095–1104. [Google Scholar] [CrossRef]
- Khateb, S.; Kowalewski, B.; Bedoni, N.; Damme, M.; Pollack, N.; Saada, A.; Obolensky, A.; Ben-Yosef, T.; Gross, M.; Dierks, T.; et al. A homozygous founder missense variant in arylsulfatase G abolishes its enzymatic activity causing atypical Usher syndrome in humans. Genet. Med. 2018, 20, 1004–1012. [Google Scholar] [CrossRef]
- Namburi, P.; Ratnapriya, R.; Khateb, S.; Lazar, C.H.; Kinarty, Y.; Obolensky, A.; Erdinest, I.; Marks-Ohana, D.; Pras, E.; Ben-Yosef, T.; et al. Bi-allelic Truncating Mutations in CEP78, Encoding Centrosomal Protein 78, Cause Cone-Rod Degeneration with Sensorineural Hearing Loss. Am. J. Hum. Genet. 2016, 99, 1222–1223. [Google Scholar] [CrossRef]
- Rizel, L.; Safieh, C.; Shalev, S.A.; Mezer, E.; Jabaly-Habib, H.; Ben-Neriah, Z.; Chervinsky, E.; Briscoe, D.; Ben-Yosef, T. Novel mutations of MYO7A and USH1G in Israeli Arab families with Usher syndrome type 1. Mol. Vis. 2011, 17, 3548–3555. [Google Scholar] [PubMed]
- Shalata, A.; Bar-Shai, M.; Hadid, Y.; Mahroum, M.; Mintz, H.; Shalata, Z.E.; Radzishevsky, E.; Genizi, J.; Lorber, A.; Ben-Yosef, T.; et al. Danon Disease: Entire LAMP2 Gene Deletion with Unusual Clinical Presentation-Case Report and Review of the Literature. Genes 2023, 14, 1539. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Ben-Yosef, T.; Goldenberg-Cohen, N.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 2020, 41, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.E.; Poulter, J.A.; Levin, A.V.; Capasso, J.E.; Price, S.; Ben-Yosef, T.; Sharony, R.; Newman, W.G.; Shore, R.C.; Brookes, S.J.; et al. Spectrum of PEX1 and PEX6 variants in Heimler syndrome. Eur. J. Hum. Genet. 2016, 24, 1565–1571. [Google Scholar] [CrossRef]
- Tatour, Y.; Sanchez-Navarro, I.; Chervinsky, E.; Hakonarson, H.; Gawi, H.; Tahsin-Swafiri, S.; Leibu, R.; Lopez-Molina, M.I.; Fernandez-Sanz, G.; Ayuso, C.; et al. Mutations in SCAPER cause autosomal recessive retinitis pigmentosa with intellectual disability. J. Med. Genet. 2017, 54, 698–704. [Google Scholar] [CrossRef]
- Ben Yosef, T.; Banin, E.; Chervinsky, E.; Shalev, S.A.; Leibu, R.; Mezer, E.; Rotenstreich, Y.; Goldenberg-Cohen, N.; Weiss, S.; Khan, M.I.; et al. Genetic causes of inherited retinal diseases among Israeli Jews of Ethiopian ancestry. Mol. Vis. 2023, 29, 1–12. [Google Scholar]
- Panneman, D.M.; Hitti-Malin, R.J.; Holtes, L.K.; de Bruijn, S.E.; Reurink, J.; Boonen, E.G.M.; Khan, M.I.; Ali, M.; Andreasson, S.; De Baere, E.; et al. Cost-effective sequence analysis of 113 genes in 1,192 probands with retinitis pigmentosa and Leber congenital amaurosis. Front. Cell Dev. Biol. 2023, 11, 1112270. [Google Scholar] [CrossRef]
- Weisschuh, N.; Feldhaus, B.; Khan, M.I.; Cremers, F.P.M.; Kohl, S.; Wissinger, B.; Zobor, D. Molecular and clinical analysis of 27 German patients with Leber congenital amaurosis. PLoS ONE 2018, 13, e0205380. [Google Scholar] [CrossRef]
- Auslender, N.; Sharon, D.; Abbasi, A.H.; Garzozi, H.J.; Banin, E.; Ben-Yosef, T. A common founder mutation of CERKL underlies autosomal recessive retinal degeneration with early macular involvement among Yemenite Jews. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5431–5438. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Fu, W.; O’Connor, T.D.; Jun, G.; Kang, H.M.; Abecasis, G.; Leal, S.M.; Gabriel, S.; Rieder, M.J.; Altshuler, D.; Shendure, J.; et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013, 493, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.C.; Gogarten, S.M.; Fullerton, S.M.; Isasi, C.R.; Mitchell, B.D.; North, K.E.; Rich, S.S.; Taylor, M.R.G.; Zollner, S.; Sofer, T. Social and scientific motivations to move beyond groups in allele frequencies: The TOPMed experience. Am. J. Hum. Genet. 2022, 109, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.M.; Halees, A.; Itan, Y.; Spencer, E.G.; He, Y.; Azab, M.A.; Gabriel, S.B.; Belkadi, A.; Boisson, B.; Abel, L.; et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 2016, 48, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, L.; Gao, H.; Padigepati, S.R.; McRae, J.F.; Li, Y.; Kosmicki, J.A.; Fritzilas, N.; Hakenberg, J.; Dutta, A.; Shon, J.; et al. Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet. 2018, 50, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.J. PERCH: A Unified Framework for Disease Gene Prioritization. Hum. Mutat. 2017, 38, 243–251. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Moles-Fernandez, A.; Duran-Lozano, L.; Montalban, G.; Bonache, S.; Lopez-Perolio, I.; Menendez, M.; Santamarina, M.; Behar, R.; Blanco, A.; Carrasco, E.; et al. Computational Tools for Splicing Defect Prediction in Breast/Ovarian Cancer Genes: How Efficient Are They at Predicting RNA Alterations? Front. Genet. 2018, 9, 366. [Google Scholar] [CrossRef]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef]
- Sprecher, E.; Bergman, R.; Richard, G.; Lurie, R.; Shalev, S.; Petronius, D.; Shalata, A.; Anbinder, Y.; Leibu, R.; Perlman, I.; et al. Hypotrichosis with juvenile macular dystrophy is caused by a mutation in CDH3, encoding P-cadherin. Nat. Genet. 2001, 29, 134–136. [Google Scholar] [CrossRef]
- Trujillano, D.; Bertoli-Avella, A.M.; Kumar Kandaswamy, K.; Weiss, M.E.; Koster, J.; Marais, A.; Paknia, O.; Schroder, R.; Garcia-Aznar, J.M.; Werber, M.; et al. Clinical exome sequencing: Results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 2017, 25, 176–182. [Google Scholar] [CrossRef]
- Yik, W.Y.; Steinberg, S.J.; Moser, A.B.; Moser, H.W.; Hacia, J.G. Identification of novel mutations and sequence variation in the Zellweger syndrome spectrum of peroxisome biogenesis disorders. Hum. Mutat. 2009, 30, E467–E480. [Google Scholar] [CrossRef]
- Raas-Rothschild, A.; Wanders, R.J.; Mooijer, P.A.; Gootjes, J.; Waterham, H.R.; Gutman, A.; Suzuki, Y.; Shimozawa, N.; Kondo, N.; Eshel, G.; et al. A PEX6-defective peroxisomal biogenesis disorder with severe phenotype in an infant, versus mild phenotype resembling Usher syndrome in the affected parents. Am. J. Hum. Genet. 2002, 70, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Bodenbender, J.P.; Marino, V.; Philipp, J.; Tropitzsch, A.; Kernstock, C.; Stingl, K.; Kempf, M.; Haack, T.B.; Zuleger, T.; Mazzola, P.; et al. Comprehensive analysis of two hotspot codons in the TUBB4B gene and associated phenotypes. Sci. Rep. 2024, 14, 10551. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.L.; Liu, X.; Wang, G.; Liu, B.; Meng, X.H.; Liu, Y. The first Chinese case with LCAEOD syndrome caused by mutation of TUBB4B gene. Int. J. Ophthalmol. 2025, 18, 753–756. [Google Scholar] [CrossRef]
- Maasz, A.; Hadzsiev, K.; Ripszam, R.; Zsigmond, A.; Maka, E.; Knezy, K.; Lesch, B.; Nemeth, A.; Bene, J.; Galik, B.; et al. TUBB4B gene mutation in Leber phenotype of congenital amaurosis syndrome associated with early-onset deafness. Eur. J. Med. Genet. 2022, 65, 104471. [Google Scholar] [CrossRef]
- McFadden, J.R.; Tolete, C.D.P.; Huang, Y.; Macnamara, E.; Sept, D.; Nesterova, G.; Gahl, W.A.; Sackett, D.L.; Malicdan, M.C.V. Clinical, genetic, and structural characterization of a novel TUBB4B tubulinopathy. Mol. Genet. Metab. Rep. 2023, 36, 100990. [Google Scholar] [CrossRef]
- Medina, G.; Perry, J.; Oza, A.; Kenna, M. Hiding in plain sight: Genetic deaf-blindness is not always Usher syndrome. Cold Spring Harb. Mol. Case Stud. 2021, 7, a006088. [Google Scholar] [CrossRef]
- Scarpato, M.; Testa, F.; Nesti, A.; Zeuli, R.; Boccia, R.; Auletta, G.; Banfi, S.; Simonelli, F.; Karali, M. A Novel Variant in TUBB4B Causes Progressive Cone-Rod Dystrophy and Early Onset Sensorineural Hearing Loss. Mol. Genet. Genom. Med. 2025, 13, e70068. [Google Scholar] [CrossRef]
- Ehrenberg, M.; Weiss, S.; Orenstein, N.; Goldenberg-Cohen, N.; Ben-Yosef, T. The co-occurrence of rare non-ocular phenotypes in patients with inherited retinal degenerations. Mol. Vis. 2019, 25, 691–702. [Google Scholar]
- Mykytyn, K.; Nishimura, D.Y.; Searby, C.C.; Shastri, M.; Yen, H.J.; Beck, J.S.; Braun, T.; Streb, L.M.; Cornier, A.S.; Cox, G.F.; et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat. Genet. 2002, 31, 435–438. [Google Scholar] [CrossRef]
- Schrauwen, I.; Helfmann, S.; Inagaki, A.; Predoehl, F.; Tabatabaiefar, M.A.; Picher, M.M.; Sommen, M.; Zazo Seco, C.; Oostrik, J.; Kremer, H.; et al. A mutation in CABP2, expressed in cochlear hair cells, causes autosomal-recessive hearing impairment. Am. J. Hum. Genet. 2012, 91, 636–645. [Google Scholar] [CrossRef]
- Weston, M.D.; Luijendijk, M.W.; Humphrey, K.D.; Moller, C.; Kimberling, W.J. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am. J. Hum. Genet. 2004, 74, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Cappuccio, G.; Sayou, C.; Tanno, P.L.; Tisserant, E.; Bruel, A.L.; Kennani, S.E.; Sa, J.; Low, K.J.; Dias, C.; Havlovicova, M.; et al. De novo SMARCA2 variants clustered outside the helicase domain cause a new recognizable syndrome with intellectual disability and blepharophimosis distinct from Nicolaides-Baraitser syndrome. Genet. Med. 2020, 22, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Van Houdt, J.K.; Nowakowska, B.A.; Sousa, S.B.; van Schaik, B.D.; Seuntjens, E.; Avonce, N.; Sifrim, A.; Abdul-Rahman, O.A.; van den Boogaard, M.J.; Bottani, A.; et al. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat. Genet. 2012, 44, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Sanders, A.A.; de Vrieze, E.; Alazami, A.M.; Alzahrani, F.; Malarkey, E.B.; Sorusch, N.; Tebbe, L.; Kuhns, S.; van Dam, T.J.; Alhashem, A.; et al. KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome. Genome Biol. 2015, 16, 293. [Google Scholar] [CrossRef]
- Cohen, B.H.; Chinnery, P.F.; Copeland, W.C. POLG-Related Disorders. In GeneReviews((R)); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2010. [Google Scholar]
- van Wijk, E.; Pennings, R.J.; te Brinke, H.; Claassen, A.; Yntema, H.G.; Hoefsloot, L.H.; Cremers, F.P.; Cremers, C.W.; Kremer, H. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am. J. Hum. Genet. 2004, 74, 738–744. [Google Scholar] [CrossRef]
- Rivolta, C.; Sweklo, E.A.; Berson, E.L.; Dryja, T.P. Missense mutation in the USH2A gene: Association with recessive retinitis pigmentosa without hearing loss. Am. J. Hum. Genet. 2000, 66, 1975–1978. [Google Scholar] [CrossRef]
- Fuster-Garcia, C.; Garcia-Garcia, G.; Jaijo, T.; Fornes, N.; Ayuso, C.; Fernandez-Burriel, M.; Sanchez-De la Morena, A.; Aller, E.; Millan, J.M. High-throughput sequencing for the molecular diagnosis of Usher syndrome reveals 42 novel mutations and consolidates CEP250 as Usher-like disease causative. Sci. Rep. 2018, 8, 17113. [Google Scholar] [CrossRef]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef]
- Aksu Uzunhan, T.; Erturk, B.; Aydin, K.; Ayaz, A.; Altunoglu, U.; Yarar, M.H.; Gezdirici, A.; Icagasioglu, D.F.; Gokpinar Ili, E.; Uyanik, B.; et al. Clinical and genetic spectrum from a prototype of ciliopathy: Joubert syndrome. Clin. Neurol. Neurosurg. 2023, 224, 107560. [Google Scholar] [CrossRef]
- Cauley, E.S.; Hamed, A.; Mohamed, I.N.; Elseed, M.; Martinez, S.; Yahia, A.; Abozar, F.; Abubakr, R.; Koko, M.; Elsayed, L.; et al. Overlap of polymicrogyria, hydrocephalus, and Joubert syndrome in a family with novel truncating mutations in ADGRG1/GPR56 and KIAA0556. Neurogenetics 2019, 20, 91–98. [Google Scholar] [CrossRef]
- Fujita, A.; Higashijima, T.; Shirozu, H.; Masuda, H.; Sonoda, M.; Tohyama, J.; Kato, M.; Nakashima, M.; Tsurusaki, Y.; Mitsuhashi, S.; et al. Pathogenic variants of DYNC2H1, KIAA0556, and PTPN11 associated with hypothalamic hamartoma. Neurology 2019, 93, e237–e251. [Google Scholar] [CrossRef] [PubMed]
- Niceta, M.; Dentici, M.L.; Ciolfi, A.; Marini, R.; Barresi, S.; Lepri, F.R.; Novelli, A.; Bertini, E.; Cappa, M.; Digilio, M.C.; et al. Co-occurrence of mutations in KIF7 and KIAA0556 in Joubert syndrome with ocular coloboma, pituitary malformation and growth hormone deficiency: A case report and literature review. BMC Pediatr. 2020, 20, 120. [Google Scholar] [CrossRef] [PubMed]
- Roosing, S.; Rosti, R.O.; Rosti, B.; de Vrieze, E.; Silhavy, J.L.; van Wijk, E.; Wakeling, E.; Gleeson, J.G. Identification of a homozygous nonsense mutation in KIAA0556 in a consanguineous family displaying Joubert syndrome. Hum. Genet. 2016, 135, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.A.; Vincent, A.L. Senior-Loken syndrome and intracranial hypertension. Ophthalmic Genet. 2020, 41, 354–357. [Google Scholar] [CrossRef]
- Banizs, B.; Pike, M.M.; Millican, C.L.; Ferguson, W.B.; Komlosi, P.; Sheetz, J.; Bell, P.D.; Schwiebert, E.M.; Yoder, B.K. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development 2005, 132, 5329–5339. [Google Scholar] [CrossRef]
- Louvi, A.; Grove, E.A. Cilia in the CNS: The quiet organelle claims center stage. Neuron 2011, 69, 1046–1060. [Google Scholar] [CrossRef]
- Francis, P.J.; Haywood, S.; Rigden, S.; Calver, D.M.; Clark, G. Benign intracranial hypertension in children following renal transplantation. Pediatr. Nephrol. 2003, 18, 1265–1269. [Google Scholar] [CrossRef]
- Zlotogora, J.; Chemke, J. Medical genetics in Israel. Eur. J. Hum. Genet. 1995, 3, 147–154. [Google Scholar] [CrossRef]
- Zeegers, M.P.; van Poppel, F.; Vlietinck, R.; Spruijt, L.; Ostrer, H. Founder mutations among the Dutch. Eur. J. Hum. Genet. 2004, 12, 591–600. [Google Scholar] [CrossRef]
- Uusimaa, J.; Kettunen, J.; Varilo, T.; Jarvela, I.; Kallijarvi, J.; Kaariainen, H.; Laine, M.; Lapatto, R.; Myllynen, P.; Niinikoski, H.; et al. The Finnish genetic heritage in 2022—From diagnosis to translational research. Dis. Models Mech. 2022, 15, dmm049490. [Google Scholar] [CrossRef]
- de Bruijn, S.E.; Rodenburg, K.; Corominas, J.; Ben-Yosef, T.; Reurink, J.; Kremer, H.; Whelan, L.; Plomp, A.S.; Berger, W.; Farrar, G.J.; et al. Optical genome mapping and revisiting short-read genome sequencing data reveal previously overlooked structural variants disrupting retinal disease-associated genes. Genet. Med. 2023, 25, 100345. [Google Scholar] [CrossRef] [PubMed]
- Nakamichi, K.; Van Gelder, R.N.; Chao, J.R.; Mustafi, D. Targeted adaptive long-read sequencing for discovery of complex phased variants in inherited retinal disease patients. Sci. Rep. 2023, 13, 8535. [Google Scholar] [CrossRef] [PubMed]
- Zeuli, R.; Karali, M.; de Bruijn, S.E.; Rodenburg, K.; Scarpato, M.; Capasso, D.; Astuti, G.D.N.; Gilissen, C.; Rodriguez-Hidalgo, M.; Ruiz-Ederra, J.; et al. Whole genome sequencing identifies elusive variants in genetically unsolved Italian inherited retinal disease patients. Hum. Genet. Genom. Adv. 2024, 5, 100314. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Family- Individual (Sex) | Origin | Age of Onset of RP (Y) | Age of Onset of HL (Y) | HL Characteristics (Age-Y) | Gene | Pathogenic Variant/s |
|---|---|---|---|---|---|---|
| TB441-1 (M) | ASH/non-Jewish | 18 | 30 | Mild (30) | USH2A | c.4174G>T; p.(Gly1392*) het |
| c.12575G>A; p.(Arg4192His) het | ||||||
| TB592-1 (M) | NAJ | 50 | 55 | Mild–moderate (59) | USH2A | c.1267-2A>G hom |
| TB639-1 (M) | OJ | 42 | 30 | Moderate (48) | USH2A | c.236_239dup; p.(Gln81Tyrfs*28) hom |
| TB699-1 (M) | NAJ | 40 | 40 | Moderate (59) | USH2A | c.1000C>T; p.(Arg334Trp) het |
| c.2167+5G>A het | ||||||
| TB785-1 (M) | MA | 54 | 57 | Mild (62) | USH2A | c.10859; p.(Ile3620Thr) hom |
| TB898-1 (M) | ASH | 70 | 18 | Moderate (76) | USH2A | c.12575G>A; p.(Arg4192His) hom |
| TB1143-1 (F) | EJ | 10 | 30 | Mild (34) | USH2A | c.784+14389G>T het |
| c.7951A>G; p.(Asn2651Asp) het | ||||||
| TB1306-1 (F) | NAJ | 20 | 40 | NA | USH2A | c.1000C>T; p.(Arg334Trp) hom |
| TB53-1 (M) | YJ | 45 | 4 | Severe–profound (63 y) | USH1C | c.1220del; p.(Gly407Glufs*58) hom |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulyamzin, S.; Leibu, R.; Newman, H.; Ehrenberg, M.; Goldenberg-Cohen, N.; Zayit-Soudry, S.; Mezer, E.; Rotenstreich, Y.; Deitch, I.; Panneman, D.M.; et al. Phenotypic and Genotypic Characterization of 171 Patients with Syndromic Inherited Retinal Diseases Highlights the Importance of Genetic Testing for Accurate Clinical Diagnosis. Genes 2025, 16, 745. https://doi.org/10.3390/genes16070745
Kulyamzin S, Leibu R, Newman H, Ehrenberg M, Goldenberg-Cohen N, Zayit-Soudry S, Mezer E, Rotenstreich Y, Deitch I, Panneman DM, et al. Phenotypic and Genotypic Characterization of 171 Patients with Syndromic Inherited Retinal Diseases Highlights the Importance of Genetic Testing for Accurate Clinical Diagnosis. Genes. 2025; 16(7):745. https://doi.org/10.3390/genes16070745
Chicago/Turabian StyleKulyamzin, Sofia, Rina Leibu, Hadas Newman, Miriam Ehrenberg, Nitza Goldenberg-Cohen, Shiri Zayit-Soudry, Eedy Mezer, Ygal Rotenstreich, Iris Deitch, Daan M. Panneman, and et al. 2025. "Phenotypic and Genotypic Characterization of 171 Patients with Syndromic Inherited Retinal Diseases Highlights the Importance of Genetic Testing for Accurate Clinical Diagnosis" Genes 16, no. 7: 745. https://doi.org/10.3390/genes16070745
APA StyleKulyamzin, S., Leibu, R., Newman, H., Ehrenberg, M., Goldenberg-Cohen, N., Zayit-Soudry, S., Mezer, E., Rotenstreich, Y., Deitch, I., Panneman, D. M., Zur, D., Chervinsky, E., Shalev, S. A., Cremers, F. P. M., Sharon, D., Roosing, S., & Ben-Yosef, T. (2025). Phenotypic and Genotypic Characterization of 171 Patients with Syndromic Inherited Retinal Diseases Highlights the Importance of Genetic Testing for Accurate Clinical Diagnosis. Genes, 16(7), 745. https://doi.org/10.3390/genes16070745