Genome Skimming Reveals Plastome Conservation, Phylogenetic Structure, and Novel Molecular Markers in Valuable Orchid Changnienia amoena


Abstract
1. Introduction
2. Results and Discussion
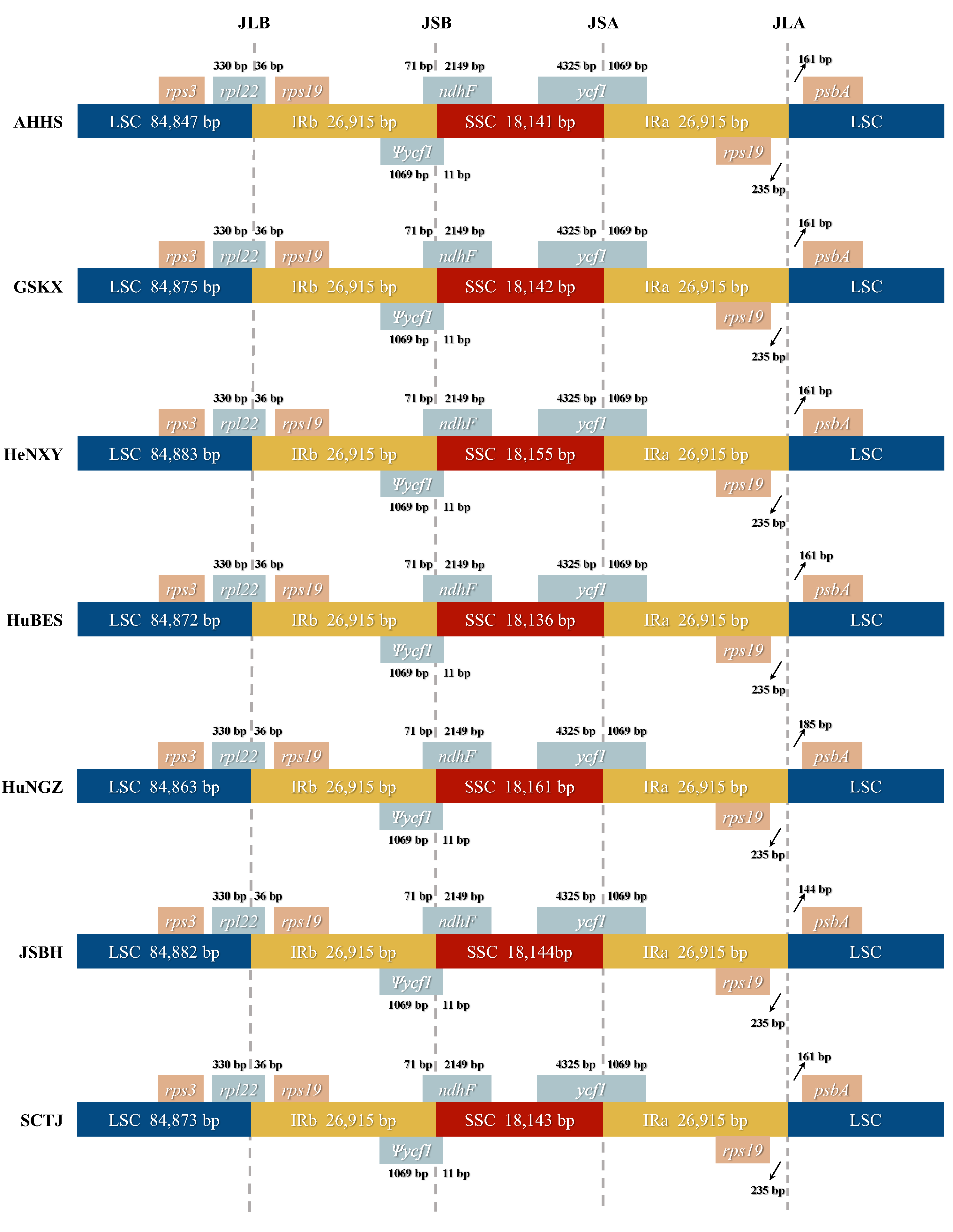
2.1. Plastome Assembly and General Features
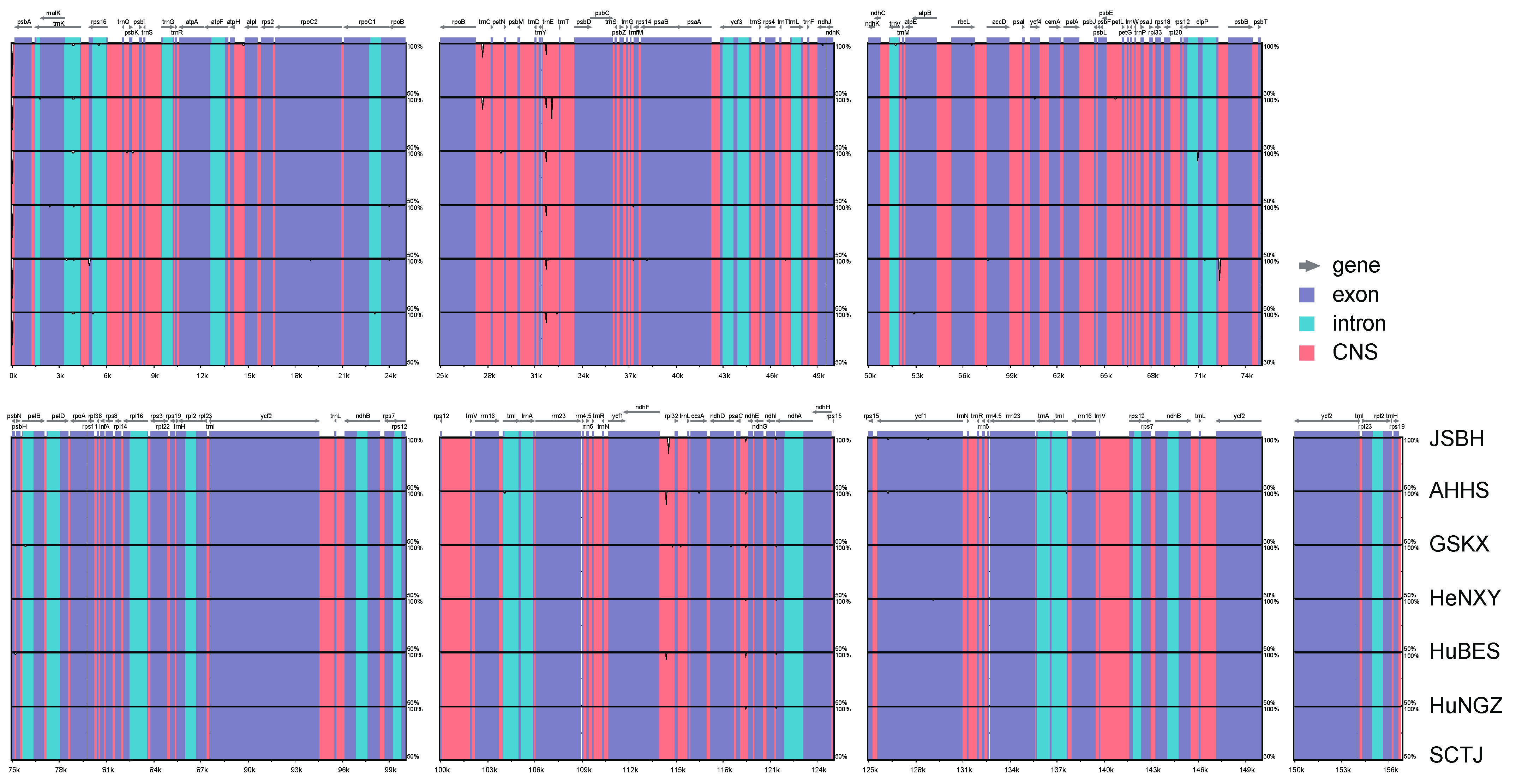
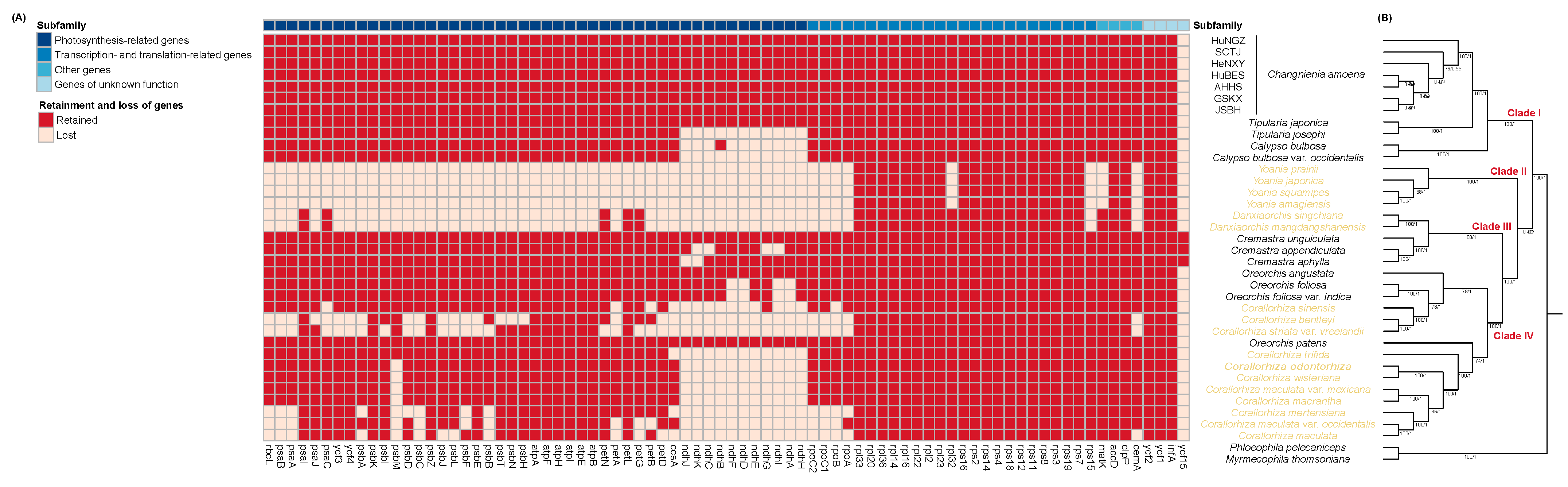
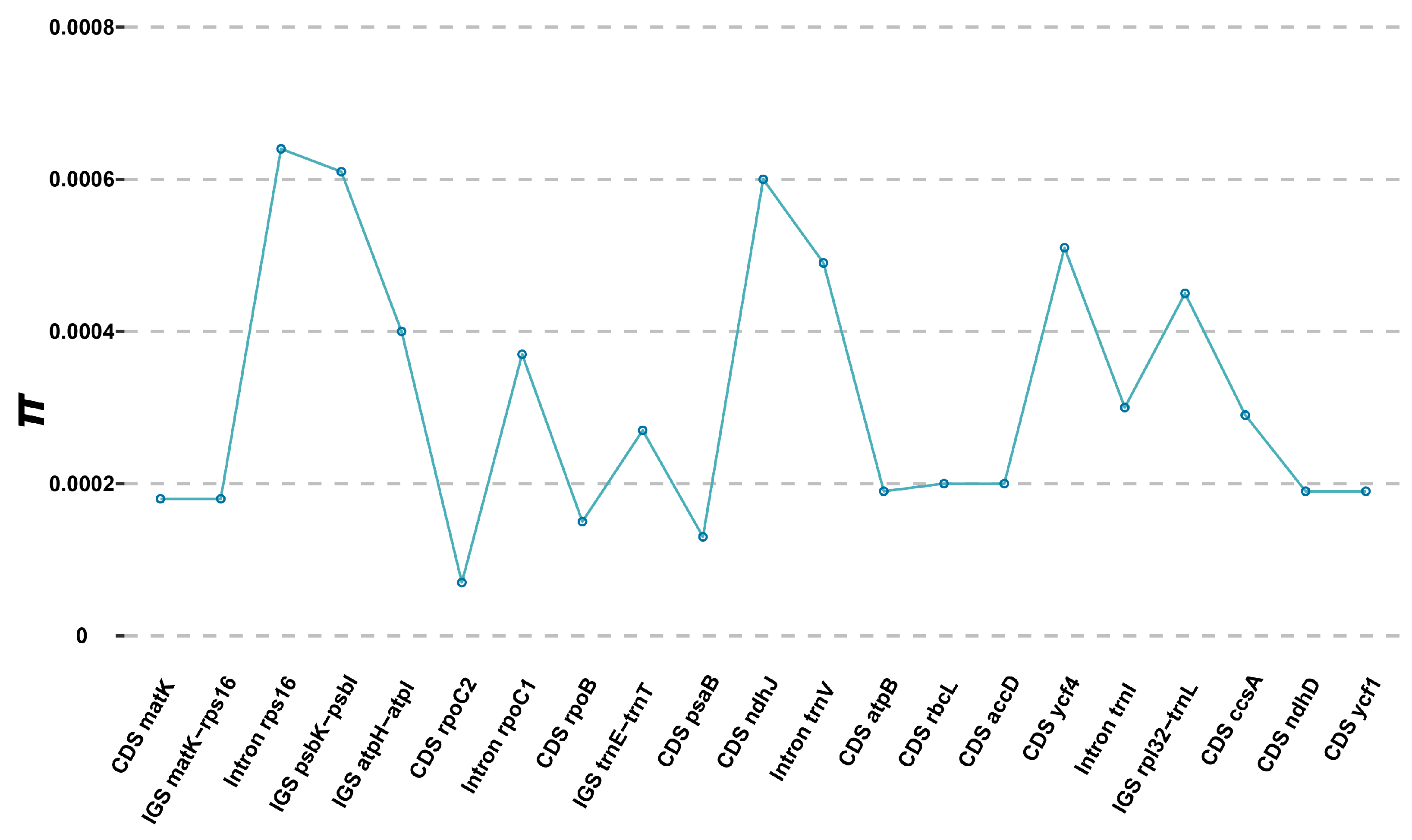
2.2. Comparative Plastome Analyses Within and Between C. amoena and Closely Related Species
2.3. Phylogenetic Relationships Both Within C. amoena and Among Calypsoinae Species
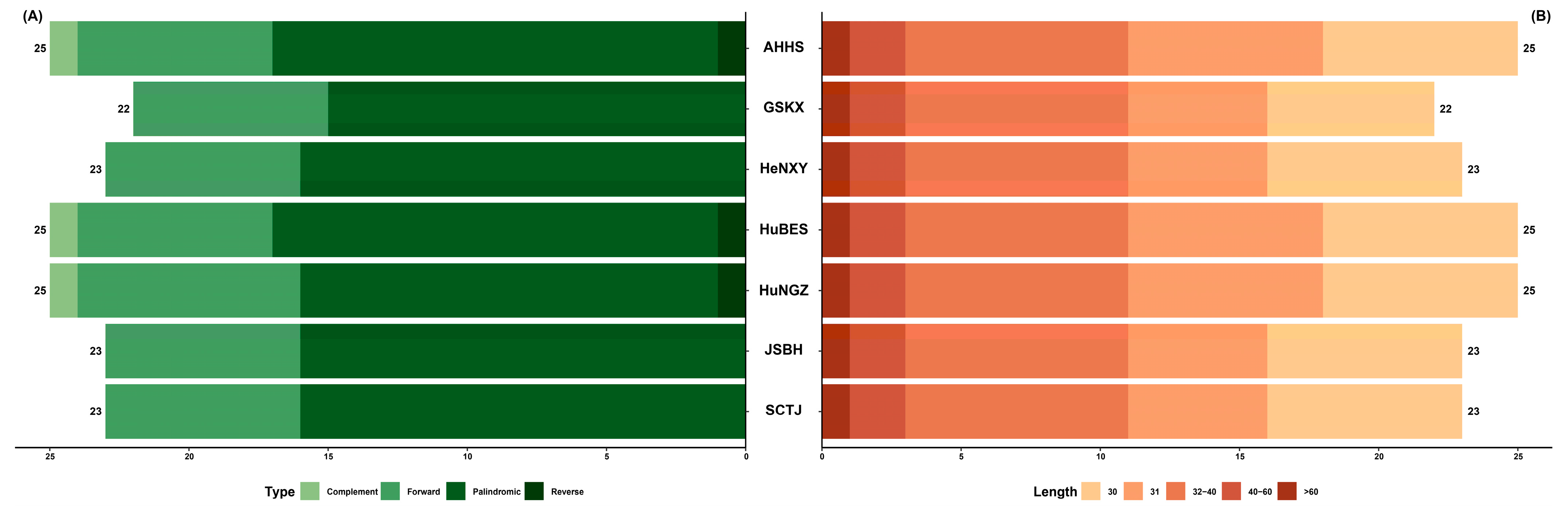
2.4. Plastome-Derived Marker Development for C. amoena
2.5. Polymorphic Nuclear SSRs for C. amoena
3. Materials and Methods
3.1. Plant Materials, DNA Extraction, and Genome Sequencing
3.2. Plastome Assembly and Annotation
3.3. Whole Plastome Sequence Comparison
3.4. Phylogenetic Analyses
3.5. Development of Plastome-Based Markers for C. amoena
3.6. Polymorphic Nuclear SSRs Development
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.J.; Wang, H.C.; Ye, C.; Wang, Z.P.; Ma, C.B.; Lin, D.L.; Jin, X.H. Progress in systematics and biogeography of Orchidaceae. Plant Divers. 2024, 46, 425–434. [Google Scholar] [CrossRef]
- Dressler, R.L. Phylogeny and Classification of the Orchid Family; Cambridge University Press: Cambridge, UK, 1993. [Google Scholar]
- Hossain, M.M.; Kant, R.; Van, P.T.; Winarto, B.; Zeng, S.; Teixeira da Silva, J.A. The application of biotechnology to orchids. Crit. Rev. Plant Sci. 2013, 32, 69–139. [Google Scholar] [CrossRef]
- Christenhusz, M.J.; Byng, J.W. The number of known plants species in the world and its annual increase. Phytotaxa 2016, 261, 201–217. [Google Scholar] [CrossRef]
- Hinsley, A.; De Boer, H.J.; Fay, M.F.; Gale, S.W.; Gardiner, L.M.; Gunasekara, R.S.; Kumar, P.; Masters, S.; Metusala, D.; Roberts, D.L.; et al. A review of the trade in orchids and its implications for conservation. Bot. J. Linn. Soc. 2018, 186, 435–455. [Google Scholar] [CrossRef]
- Choudhary, D.; Mashkey, V.K.; Goutam, E.; Shrivastava, M.; Rawat, M.; Kumari, A.; Tripathi, V. Medicinal orchids: Traditional uses and recent advances. Ann. Phytomed. 2023, 12, 1–9. [Google Scholar] [CrossRef]
- Lüning, B. Alkaloid content of Orchidaceae. In The Orchids Scientific Studies; Withner, C.L., Ed.; John Wiley and Sons: New York, NY, USA, 1974. [Google Scholar]
- Kimura, K. New species of Dendrobium from the Chinese drug Shih-hu. J. Shanghai Sci. Inst. Sect. III 1936, 3, 121–124. [Google Scholar]
- Kaushik, P. Ecological and Anatomical Marvels of the Himalayan Orchids; Today and Tomorrow’s Printers and Publishers: New Delhi, India, 1983; Volume 8. [Google Scholar]
- Zhang, S.B.; Yang, Y.J.; Li, J.W.; Qin, J.; Zhang, W.; Huang, W.; Hu, H. Physiological diversity of orchids. Plant Divers. 2018, 40, 196–208. [Google Scholar] [CrossRef]
- Chase, M.W. Classification of Orchidaceae in the age of DNA data. Curtis’s Bot. Mag. 2005, 22, 2–7. [Google Scholar] [CrossRef]
- Silvera, K.; Santiago, L.S.; Cushman, J.C.; Winter, K. Crassulacean acid metabolism and epiphytism linked to adaptive radiations in the Orchidaceae. Plant Physiol. 2009, 149, 1838–1847. [Google Scholar] [CrossRef]
- Givnish, T.J.; Spalink, D.; Ames, M.; Lyon, S.P.; Hunter, S.J.; Zuluaga, A.; Iles, W.J.; Clements, M.A.; Arroyo, M.T.; Leebens-Mack, J.; et al. Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proc. R. Soc. B 2015, 282, 20151553. [Google Scholar] [CrossRef]
- Li, A.; Ge, S. Genetic variation and conservation of Changnienia amoena, an endangered orchid endemic to China. Plant Syst. Evol. 2006, 258, 251–260. [Google Scholar] [CrossRef]
- Fay, M.F.; Chase, M.W. Orchid biology: From Linnaeus via Darwin to the 21st century. Ann. Bot. 2009, 104, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Chin, C.M. China Plant Red Data Book; Science Press: Alexandria, Australia, 1992. [Google Scholar]
- Xiong, G.M.; Xie, Z.Q.; Xiong, X.G.; Fan, D.Y.; Ge, S. The biology and community characteristics of Changnienia amoena distributed in southern part of Shennongjia region. Acta Ecol. Sin. 2003, 23, 187–194. [Google Scholar]
- Jiang, W.M.; Yang, G.M.; Zhang, C.L.; Fu, C.X. Species composition and molecular analysis of symbiotic fungi in roots of Changnienia amoena (Orchidaceae). Afr. J. Microbiol. Res. 2011, 5, 222–228. [Google Scholar]
- Sun, H.Q.; Luo, Y.B.; Alexandersson, R.; Ge, S. Pollination biology of the deceptive orchid Changnienia amoena. Bot. J. Linn. Soc. 2006, 150, 165–175. [Google Scholar] [CrossRef]
- Liu, M.X.; Wang, X.W.; Yang, C.L. Exploring the potential distribution areas of Changnienia amoena and its pollinators in China based on MaxEnt and GTWR models. J. Nat. Conserv. 2025, 86, 126946. [Google Scholar] [CrossRef]
- Chen, X.Q.; Ji, Z.H. The Orchids of China; Chinese Forestry Publisher: Beijing, China, 1998. [Google Scholar]
- Wang, N.H.; Lu, Y.; Cheng, Z.L. Observation of biological properties and preliminary study on reproduction of Changnienia amoena Chien. Chin. Bull. Bot. 1994, 11, 53–55. [Google Scholar]
- Yi, X.G.; Li, M.Z.; Chen, L.; Wang, X.R. The complete chloroplast genome of Changnienia amoena SS Chien (Orchidaceae) and its phylogenetic implication. Mitochondrial DNA Part B 2020, 5, 238–239. [Google Scholar] [CrossRef]
- Dodsworth, S. Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 2015, 20, 525–527. [Google Scholar] [CrossRef]
- Straub, S.C.; Parks, M.; Weitemier, K.; Fishbein, M.; Cronn, R.C.; Liston, A. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 2012, 99, 349–364. [Google Scholar] [CrossRef]
- Birky Jr, C.W.; Maruyama, T.; Fuerst, P. An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics 1983, 103, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Chen, M.; Li, P.; Sun, X.Q.; Lu, R.S. Intraspecific phylogeny and genomic resources development for an important medical plant Dioscorea nipponica, based on low-coverage whole genome sequencing data. Front. Plant Sci. 2023, 14, 1320473. [Google Scholar] [CrossRef]
- Lu, R.S.; Hu, K.; Sun, X.Q.; Chen, M. Low-coverage whole genome sequencing of diverse Dioscorea bulbifera accessions for plastome resource development, polymorphic nuclear SSR identification, and phylogenetic analyses. Front. Plant Sci. 2024, 15, 1373297. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.S.; Yang, T.; Chen, Y.; Wang, S.Y.; Cai, M.Q.; Cameron, K.M.; Li, P.; Fu, C.X. Comparative plastome genomics and phylogenetic analyses of Liliaceae. Bot. J. Linn. Soc. 2021, 196, 279–293. [Google Scholar] [CrossRef]
- Chen, X.Y.; Xiang, X.G.; Liu, X.D.; Li, W.Y.; Wu, X.C.; Zhou, Y.D.; Yang, B.Y.; Luo, H.L. Comparison of chloroplast genomes of Calypsoinae species (Orchidaceae) living on different lifeforms. Res. Sq. 2023. preprint. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef]
- Lu, R.S.; Chen, M.; Feng, Y.; Yuan, N.; Zhang, Y.M.; Cao, M.X.; Liu, J.; Wang, Y.; Hang, Y.Y.; Sun, X.Q. Comparative plastome analyses and genomic resource development in wild rice (Zizania spp., Poaceae) using genome skimming data. Ind. Crops Prod. 2022, 186, 115244. [Google Scholar] [CrossRef]
- Li, Z.H.; Jiang, Y.; Ma, X.; Li, J.W.; Yang, J.B.; Wu, J.Y.; Jin, X.H. Plastid Genome Evolution in the Subtribe Calypsoinae (Epidendroideae, Orchidaceae). Genome Biol. Evol. 2020, 12, 867–870. [Google Scholar] [CrossRef]
- Yang, J.X.; Peng, S.; Wang, J.J.; Ding, S.X.; Wang, Y.; Tian, J.; Yang, H.; Hu, G.W.; Wang, Q.F. Morphological and genomic evidence for a new species of Corallorhiza (Orchidaceae: Epidendroideae) from SW China. Plant Divers. 2021, 43, 409–419. [Google Scholar] [CrossRef]
- Lee, S.Y.; Meng, K.K.; Wang, H.W.; Zhou, R.C.; Liao, W.B.; Chen, F.; Zhang, S.Z.; Fan, Q. Severe plastid genome size reduction in a mycoheterotrophic orchid, Danxiaorchis singchiana, reveals heavy gene loss and gene relocations. Plants 2020, 9, 521. [Google Scholar] [CrossRef]
- Barrett, C.F.; Davis, J.I. The plastid genome of the mycoheterotrophic Corallorhiza striata (Orchidaceae) is in the relatively early stages of degradation. Am. J. Bot. 2012, 99, 1513–1523. [Google Scholar] [CrossRef]
- Ferdy, J.B.; Loriot, S.; Sandmeier, M.; Lefranc, M.; Raquin, C. Inbreeding depression in a rare deceptive orchid. Can. J. Bot. 2001, 79, 1181–1188. [Google Scholar]
- Barrett, C.F.; Freudenstein, J.V.; Skibicki, S.V.; Sinn, B.T.; Chung, S.W.; Hsu, T.C.; Liao, W.; Lee, S.Y.; Luo, Y.B.; Yukawa, T.; et al. Phylogenomics and intergenomic conflict in a challenging orchid clade (Calypsoinae): Monophyly of Corallorhiza, paraphyly of Oreorchis, and resurrection of Kitigorchis. Bot. J. Linn. Soc. 2025, boae092. [Google Scholar] [CrossRef]
- Freudenstein, J.V.; Yukawa, T.; Luo, Y.B. A reanalysis of relationships among Calypsoinae (Orchidaceae: Epidendroideae): Floral and vegetative evolution and the placement of Yoania. Syst. Bot. 2017, 42, 17–25. [Google Scholar] [CrossRef]
- Liu, Z.; Lee, S.Y.; Yeh, C.L.; Averyanov, L.V.; Liao, W.; Suetsugu, K. Plastome analysis elucidates the phylogenetic placement of the mycoheterotrophic genus Yoania (Orchidaceae) and its plastomic degeneration during the evolution of mycoheterotrophy. Bot. J. Linn. Soc. 2024, 206, 313–326. [Google Scholar] [CrossRef]
- Gu, C.H.; Tembrock, L.R.; Zheng, S.Y.; Wu, Z.Q. The complete chloroplast genome of Catha edulis: A comparative analysis of genome features with related species. Int. J. Mol. Sci. 2018, 19, 525. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Liu, J.; Yu, J.; Wang, L.; Zhou, S.L. Highly Variable Chloroplast Markers for Evaluating Plant Phylogeny at Low Taxonomic Levels and for DNA Barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef]
- Provan, P.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Zhou, Q.Y.; Cai, H.X.; Liu, Z.H.; Yuan, L.X.; Yang, L.; Yang, T.; Li, B.; Li, P. Development of genomic resources for Wenchengia alternifolia (Lamiaceae) based on genome skimming data. Plant Divers. 2022, 44, 542–551. [Google Scholar] [CrossRef]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Zhang, S.Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef]
- Li, H.M.; Wu, M.S.; Lai, Q.; Zhou, W.; Song, C.F. Complete chloroplast of four Sanicula taxa (Apiaceae) endemic to China: Lights into genome structure, comparative analysis, and phylogenetic relationships. BMC Plant Biol. 2023, 23, 444. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Zhu, H.H.; Wang, J.; Xu, Y.C.; Xu, F.S.; Wang, X.M. Complete chloroplast genome sequence determination of Rheum species and comparative chloroplast genomics for the members of Rumiceae. Plant Cell Rep. 2020, 39, 811–824. [Google Scholar] [CrossRef]
- Liang, H.; Zhang, Y.; Deng, J.B.; Gao, G.; Ding, C.B.; Zhang, L.; Yang, R.W. The Complete Chloroplast Genome Sequences of 14 Curcuma Species: Insights into Genome Evolution and Phylogenetic Relationships Within Zingiberales. Front. Genet. 2020, 11, 802. [Google Scholar] [CrossRef]
- Smidt, E.d.C.; Páez, M.Z.; Vieira, L.d.N.; Viruel, J.; de Baura, V.A.; Balsanelli, E.; de Souza, E.M.; Chase, M.W. Characterization of sequence variability hotspots in Cranichideae plastomes (Orchidaceae, Orchidoideae). PLoS ONE 2020, 15, e0227991. [Google Scholar] [CrossRef]
- Kim, Y.K.; Jo, S.; Cheon, S.H.; Joo, M.J.; Hong, J.R.; Kwak, M.; Kim, K.J. Plastome evolution and phylogeny of Orchidaceae, with 24 new sequences. Front. Plant Sci. 2020, 11, 22. [Google Scholar] [CrossRef]
- Hollingsworth, P.M.; Li, D.Z.; van der Bank, M.; Twyford, A.D. Telling plant species apart with DNA: From barcodes to genomes. Philos. Trans. R. Soc. B 2016, 371, 20150338. [Google Scholar] [CrossRef] [PubMed]
- Aecyo, P.; Marques, A.; Huettel, B.; Silva, A.; Esposito, T.; Ribeiro, E.; Leal, I.R.; Gagnon, E.; Souza, G.; Pedrosa-Harand, A. Plastome evolution in the Caesalpinia group (Leguminosae) and its application in phylogenomics and populations genetics. Planta 2021, 254, 27. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; DePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; Green, E.D.; Sidow, A.; Batzoglou, S.; Program, N.C.S. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32 (Suppl. S2), W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar]
- Li, R.Q.; Zhu, H.M.; Ruan, J.; Qian, W.B.; Fang, X.D.; Shi, Z.B.; Li, Y.R.; Li, S.T.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Xia, E.H.; Yao, Q.Y.; Zhang, H.B.; Jiang, J.J.; Zhang, L.P.; Gao, L.Z. CandiSSR: An efficient pipeline used for identifying candidate polymorphic SSRs based on multiple assembled sequences. Front. Plant Sci. 2016, 6, 1171. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | AHHS | GSKX | HeNXY | HuBES | HuNGZ | JSBH | SCTJ |
|---|---|---|---|---|---|---|---|
| Locality | Huoshan, Anhui | Kangxian, Gansu | Xinyang, Henan | Enshi, Hubei | Guzhang, Hunan | Baohua, Jiangsu | Tongjiang, Sichuan |
| Plastome Size (bp) | 156,854 | 156,868 | 156,856 | 156,847 | 156,838 | 156,818 | 156,846 |
| Average base coverage (×) | 429.3 | 351.2 | 317.8 | 244.9 | 171.4 | 363.2 | 399.2 |
| LSC length (bp) | 84,863 | 84,883 | 84,882 | 84,875 | 84,872 | 84,847 | 84,873 |
| SSC length (bp) | 18,161 | 18,155 | 18,144 | 18,142 | 18,136 | 18,141 | 18,143 |
| IR length (bp) | 26,915 | 26,915 | 26,915 | 26,915 | 26,915 | 26,915 | 26,915 |
| GC content (%) | 37.10% | 37.10% | 37.10% | 37.10% | 37.10% | 37.10% | 37.10% |
| LSC GC content (%) | 34.80% | 34.80% | 34.80% | 34.80% | 34.80% | 34.80% | 34.80% |
| SSC GC content (%) | 29.80% | 29.80% | 29.80% | 29.80% | 29.80% | 29.80% | 29.80% |
| IR GC content (%) | 43.20% | 43.20% | 43.20% | 43.20% | 43.20% | 43.20% | 43.20% |
| Total genes | 113 | 113 | 113 | 113 | 113 | 113 | 113 |
| Total PCGs | 87 | 87 | 87 | 87 | 87 | 87 | 87 |
| Total tRNAs | 38 | 38 | 38 | 38 | 38 | 38 | 38 |
| Total rRNAs | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Duplicated genes | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| GenBank accession number | PV612366 | PV612364 | PV612367 | PV612363 | PV612365 | PV612362 | PV612368 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, R.-S.; Hu, K.; Liu, Y.; Sun, X.-Q.; Liu, X.-J. Genome Skimming Reveals Plastome Conservation, Phylogenetic Structure, and Novel Molecular Markers in Valuable Orchid Changnienia amoena. Genes 2025, 16, 723. https://doi.org/10.3390/genes16070723
Lu R-S, Hu K, Liu Y, Sun X-Q, Liu X-J. Genome Skimming Reveals Plastome Conservation, Phylogenetic Structure, and Novel Molecular Markers in Valuable Orchid Changnienia amoena. Genes. 2025; 16(7):723. https://doi.org/10.3390/genes16070723
Chicago/Turabian StyleLu, Rui-Sen, Ke Hu, Yu Liu, Xiao-Qin Sun, and Xing-Jian Liu. 2025. "Genome Skimming Reveals Plastome Conservation, Phylogenetic Structure, and Novel Molecular Markers in Valuable Orchid Changnienia amoena" Genes 16, no. 7: 723. https://doi.org/10.3390/genes16070723
APA StyleLu, R.-S., Hu, K., Liu, Y., Sun, X.-Q., & Liu, X.-J. (2025). Genome Skimming Reveals Plastome Conservation, Phylogenetic Structure, and Novel Molecular Markers in Valuable Orchid Changnienia amoena. Genes, 16(7), 723. https://doi.org/10.3390/genes16070723