
Expansion of the Genotypic and Phenotypic Spectrum of TCTN3-Related Joubert Syndrome

 , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Examination
2.2. Next-Generation Sequencing Analysis
2.3. Sanger Sequencing
2.4. cDNA Analysis
3. Results
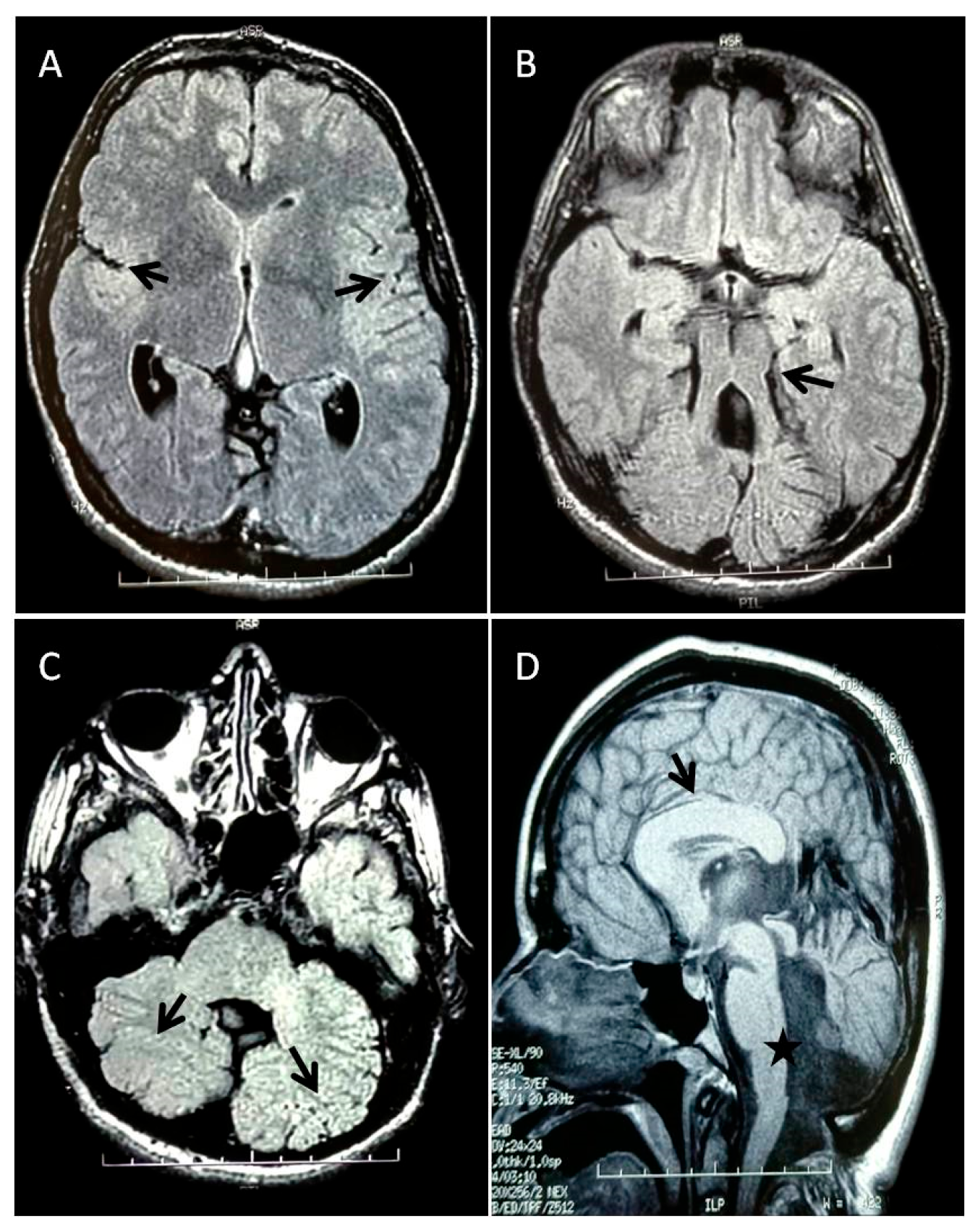
3.1. Patient’s Clinical History
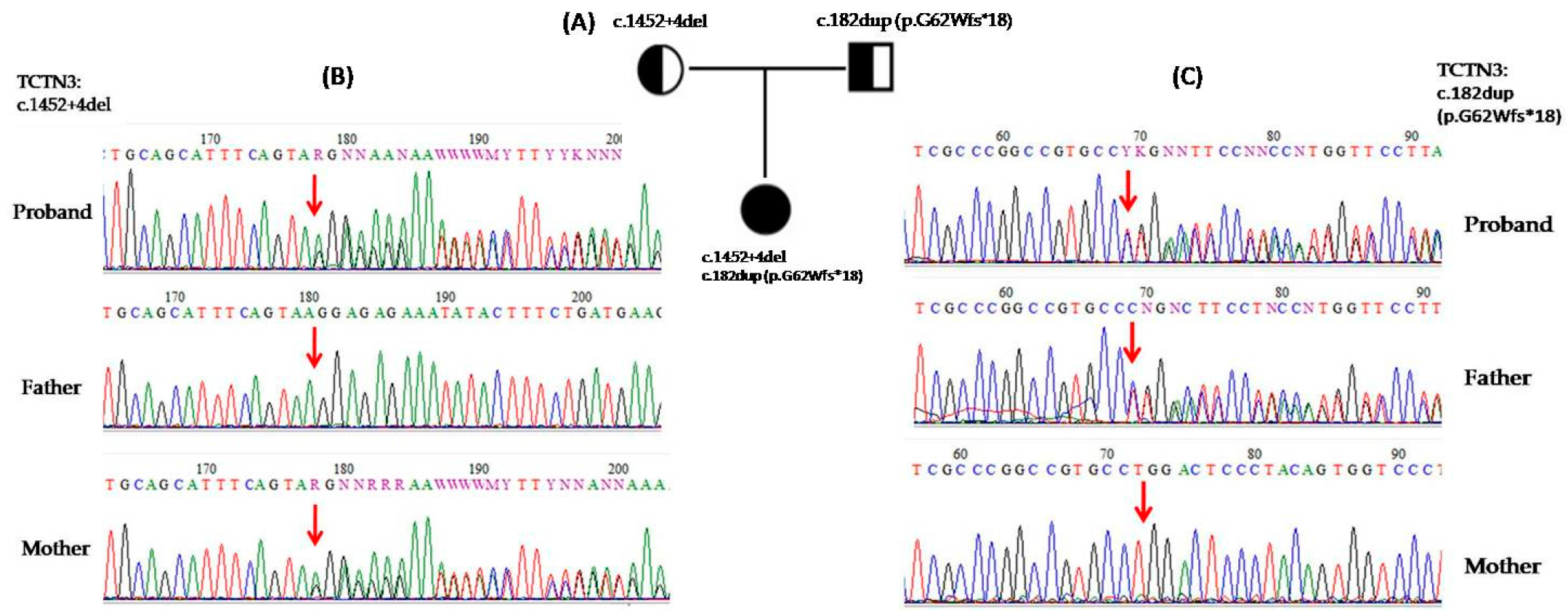
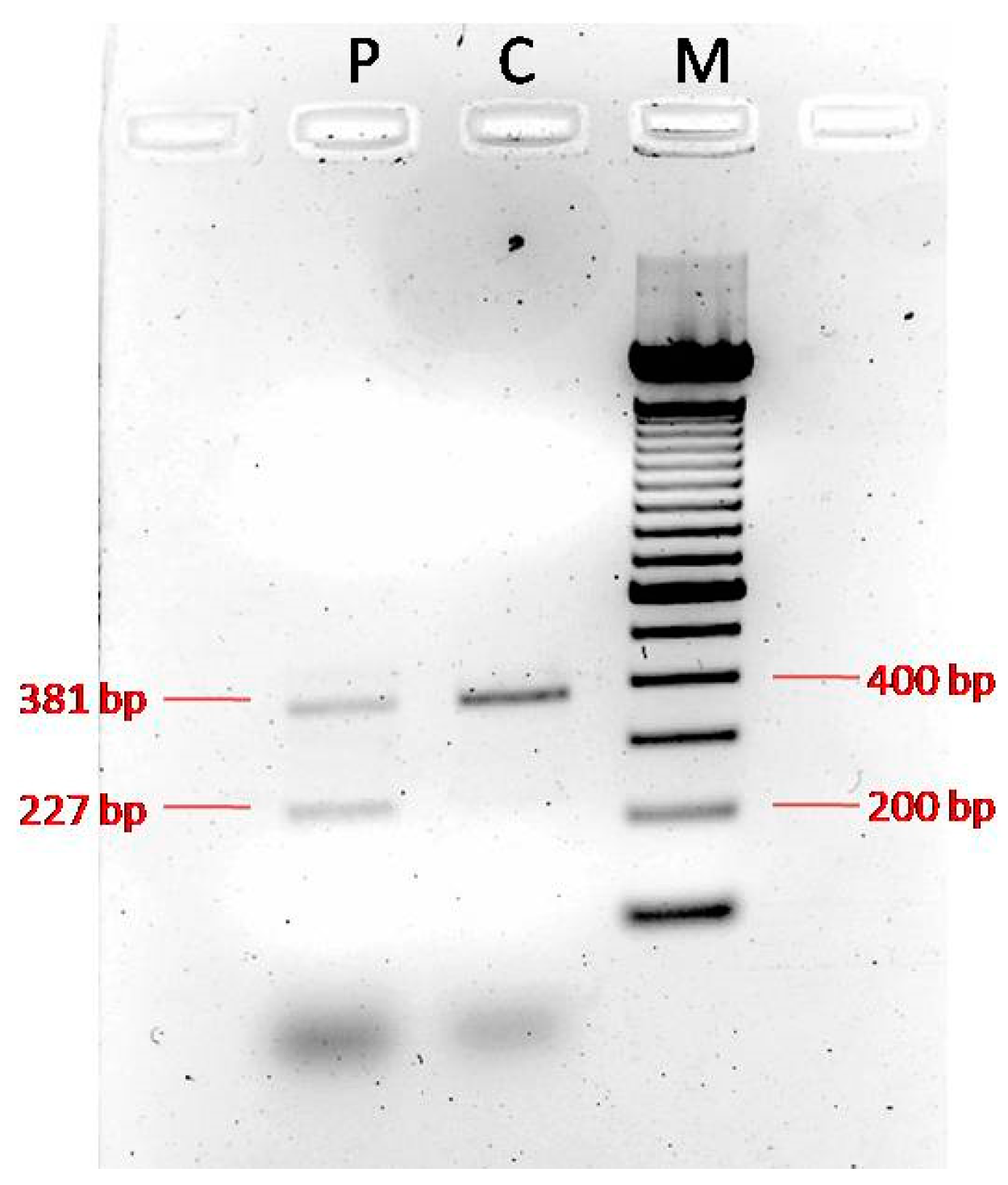
3.2. Genetic and Functional Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| JS | Joubert syndrome |
| MTS | Molar tooth sign |
| MRI | Magnetic resonance imaging |
| CNS | Central nervous system |
| MKS | Meckel–Gruber Syndrome |
| OFD | Orofaciodigital syndrome IV |
| ID | Intellectual disability |
| WES | Whole-exome sequencing |
| EEG | Electroencephalogram |
| ACC | Agenesis of the corpus callosum |
| MCAP | Megalencephaly-Capillary Malformation Syndrome |
References
- Joubert, M.; Eisenring, J.J.; Robb, J.P.; Andermann, F. Familial Agenesis of the Cerebellar Vermis. A Syndrome of Episodic Hyperpnea, Abnormal Eye Movements, Ataxia, and Retardation. Neurology 1969, 19, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Dallapiccola, B.; Bertini, E. Joubert Syndrome and Related Disorders. Handb. Clin. Neurol. 2013, 113, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, J.G.; Keeler, L.C.; Parisi, M.A.; Marsh, S.E.; Chance, P.F.; Glass, I.A.; Graham, J.M.; Maria, B.L.; Barkovich, A.J.; Dobyns, W.B. Molar Tooth Sign of the Midbrain-Hindbrain Junction: Occurrence in Multiple Distinct Syndromes. Am. J. Med. Genet. A 2004, 125A, 125–134, discussion 117. [Google Scholar] [CrossRef] [PubMed]
- Bachmann-Gagescu, R.; Dempsey, J.C.; Phelps, I.G.; O’Roak, B.J.; Knutzen, D.M.; Rue, T.C.; Ishak, G.E.; Isabella, C.R.; Gorden, N.; Adkins, J.; et al. Joubert Syndrome: A Model for Untangling Recessive Disorders with Extreme Genetic Heterogeneity. J. Med. Genet. 2015, 52, 514–522. [Google Scholar] [CrossRef]
- Poretti, A.; Huisman, T.a.G.M.; Scheer, I.; Boltshauser, E. Joubert Syndrome and Related Disorders: Spectrum of Neuroimaging Findings in 75 Patients. AJNR Am. J. Neuroradiol. 2011, 32, 1459–1463. [Google Scholar] [CrossRef]
- Braddock, B.A.; Farmer, J.E.; Deidrick, K.M.; Iverson, J.M.; Maria, B.L. Oromotor and Communication Findings in Joubert Syndrome: Further Evidence of Multisystem Apraxia. J. Child. Neurol. 2006, 21, 160–163. [Google Scholar] [CrossRef]
- Brancati, F.; Dallapiccola, B.; Valente, E.M. Joubert Syndrome and Related Disorders. Orphanet J. Rare Dis. 2010, 5, 20. [Google Scholar] [CrossRef]
- Szymanska, K.; Hartill, V.L.; Johnson, C.A. Unraveling the Genetics of Joubert and Meckel-Gruber Syndromes. J. Pediatr. Genet. 2014, 3, 65–78. [Google Scholar] [CrossRef]
- Valente, E.M.; Rosti, R.O.; Gibbs, E.; Gleeson, J.G. Primary Cilia in Neurodevelopmental Disorders. Nat. Rev. Neurol. 2014, 10, 27–36. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef]
- Gana, S.; Serpieri, V.; Valente, E.M. Genotype-Phenotype Correlates in Joubert Syndrome: A Review. Am. J. Med. Genet. C Semin. Med. Genet. 2022, 190, 72–88. [Google Scholar] [CrossRef] [PubMed]
- Spahiu, L.; Behluli, E.; Grajçevci-Uka, V.; Liehr, T.; Temaj, G. Joubert Syndrome: Molecular Basis and Treatment. J. Mother Child 2023, 26, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Linpeng, S.; Liu, J.; Pan, J.; Cao, Y.; Teng, Y.; Liang, D.; Li, Z.; Wu, L. Diagnosis of Joubert Syndrome 10 in a Fetus with Suspected Dandy-Walker Variant by WES: A Novel Splicing Mutation in OFD1. Biomed. Res. Int. 2018, 2018, 4032543. [Google Scholar] [CrossRef] [PubMed]
- Chaki, M.; Airik, R.; Ghosh, A.K.; Giles, R.H.; Chen, R.; Slaats, G.G.; Wang, H.; Hurd, T.W.; Zhou, W.; Cluckey, A.; et al. Exome Capture Reveals ZNF423 and CEP164 Mutations, Linking Renal Ciliopathies to DNA Damage Response Signaling. Cell 2012, 150, 533–548. [Google Scholar] [CrossRef]
- Nuovo, S.; Bacigalupo, I.; Ginevrino, M.; Battini, R.; Bertini, E.; Borgatti, R.; Casella, A.; Micalizzi, A.; Nardella, M.; Romaniello, R.; et al. Age and Sex Prevalence Estimate of Joubert Syndrome in Italy. Neurology 2020, 94, e797–e801. [Google Scholar] [CrossRef]
- Reiter, J.F.; Skarnes, W.C. Tectonic, a Novel Regulator of the Hedgehog Pathway Required for Both Activation and Inhibition. Genes Dev. 2006, 20, 22–27. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, Y.; Dong, H.; Gong, S.; Wei, B.; Luo, M.; Wang, H.; Wu, X.; Liu, W.; Xu, X.; et al. Loss of Tctn3 Causes Neuronal Apoptosis and Neural Tube Defects in Mice. Cell Death Dis. 2018, 9, 520. [Google Scholar] [CrossRef]
- Thomas, S.; Legendre, M.; Saunier, S.; Bessières, B.; Alby, C.; Bonnière, M.; Toutain, A.; Loeuillet, L.; Szymanska, K.; Jossic, F.; et al. TCTN3 Mutations Cause Mohr-Majewski Syndrome. Am. J. Hum. Genet. 2012, 91, 372–378. [Google Scholar] [CrossRef]
- Ben-Salem, S.; Al-Shamsi, A.M.; Gleeson, J.G.; Ali, B.R.; Al-Gazali, L. Mutation Spectrum of Joubert Syndrome and Related Disorders among Arabs. Hum. Genome Var. 2014, 1, 14020. [Google Scholar] [CrossRef]
- Huppke, P.; Wegener, E.; Böhrer-Rabel, H.; Bolz, H.J.; Zoll, B.; Gärtner, J.; Bergmann, C. Tectonic Gene Mutations in Patients with Joubert Syndrome. Eur. J. Hum. Genet. 2015, 23, 616–620. [Google Scholar] [CrossRef]
- Huang, L.-X.; Lu, X.-G.; Liu, J.-X.; Xu, L.; Shang, N.; Guo, L.; OuYang, Y.-C. Case Report and a Brief Review: Analysis and Challenges of Prenatal Imaging Phenotypes and Genotypes in Joubert Syndrome. Front. Genet. 2022, 13, 1038274. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Nawaz, S.; Khan, H.; Acharya, A.; Schrauwen, I.; Ahmad, W.; Leal, S.M. A Splice Site Variant in TCTN3 Underlies an Atypical Form of Orofaciodigital Syndrome IV. Ann. Hum. Genet. 2022, 86, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Turkyilmaz, A.; Geckinli, B.B.; Alavanda, C.; Arslan Ates, E.; Buyukbayrak, E.E.; Eren, S.F.; Arman, A. Meckel-Gruber Syndrome: Clinical and Molecular Genetic Profiles in Two Fetuses and Review of the Current Literature. Genet. Test. Mol. Biomark. 2021, 25, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Mirzaa, G.M.; Conway, R.L.; Gripp, K.W.; Lerman-Sagie, T.; Siegel, D.H.; deVries, L.S.; Lev, D.; Kramer, N.; Hopkins, E.; Graham, J.M., Jr.; et al. Megalencephaly-Capillary Malformation (MCAP) and Megalencephaly-Polydactyly-Polymicrogyria-Hydrocephalus (MPPH) Syndromes: Two Closely Related Disorders of Brain Overgrowth and Abnormal Brain and Body Morphogenesis. Am. J. Med. Genet. Part A 2012, 158A, 269–291. [Google Scholar] [CrossRef]
- Sarma, K.; Nayak, M.K.; Mishra, B.; Gaikwad, S.B. Megalencephaly-Capillary Malformation-Polymicrogyria Syndrome (MCAP): A Rare Dynamic Genetic Disorder. Cureus 2022, 14, e25123. [Google Scholar] [CrossRef]
- Nguyen, L.S.; Schneider, T.; Rio, M.; Moutton, S.; Siquier-Pernet, K.; Verny, F.; Boddaert, N.; Desguerre, I.; Munich, A.; Rosa, J.L.; et al. A Nonsense Variant in HERC1 Is Associated with Intellectual Disability, Megalencephaly, Thick Corpus Callosum and Cerebellar Atrophy. Eur. J. Hum. Genet. 2016, 24, 455–458. [Google Scholar] [CrossRef]
- Gong, S.; Ji, F.; Wang, B.; Zhang, Y.; Xu, X.; Sun, M. Tectonic Proteins Are Important Players in Non-Motile Ciliopathies. Cell Physiol. Biochem. 2018, 50, 398–409. [Google Scholar] [CrossRef]
- Huljev Frković, S.; Vičić, A.; Crkvenac Gornik, K.; Kulišić, D.; Stipoljev, F. Prenatally Detected Encephalocele Associated with a Novel Pathogenic TCTN3 Variant: A Case Report and Literature Review. Am. J. Med. Genet. A 2022, 188, 1826–1830. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| c.182dup (p.G62Wfs*18) | ||
|---|---|---|
| Criteria for Classifying Variants | Category Code | Description |
| Pathogenic Very Strong: | PVS1 | Null variant in a gene where loss of function is a known mechanism of disease |
| Pathogenic Moderate: | PM2 | Extremely low frequency in gnomAD population databases |
| Pathogenic Supporting: | PP5 | A reputable source recently reported the variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation |
| ACMG variant classification | Pathogenic | |
| c.1452+4del | ||
|---|---|---|
| Criteria for Classifying Variants | Category Code | Description |
| Pathogenic Moderate: | PP3 | For a missense or a splicing region variant, computational prediction tools unanimously support a deleterious effect on the gene |
| Pathogenic Moderate: | PM2 | Extremely low frequency in gnomAD population databases |
| ACMG variant classification | Uncertain significance | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo Giudice, M.; Borgione, E.; Giuliano, M.; Santa Paola, S.; Di Blasi, F.D.; Pettinato, R.; Romano, C.; Scuderi, C. Expansion of the Genotypic and Phenotypic Spectrum of TCTN3-Related Joubert Syndrome. Genes 2025, 16, 706. https://doi.org/10.3390/genes16060706
Lo Giudice M, Borgione E, Giuliano M, Santa Paola S, Di Blasi FD, Pettinato R, Romano C, Scuderi C. Expansion of the Genotypic and Phenotypic Spectrum of TCTN3-Related Joubert Syndrome. Genes. 2025; 16(6):706. https://doi.org/10.3390/genes16060706
Chicago/Turabian StyleLo Giudice, Mariangela, Eugenia Borgione, Marika Giuliano, Sandro Santa Paola, Francesco Domenico Di Blasi, Rosa Pettinato, Corrado Romano, and Carmela Scuderi. 2025. "Expansion of the Genotypic and Phenotypic Spectrum of TCTN3-Related Joubert Syndrome" Genes 16, no. 6: 706. https://doi.org/10.3390/genes16060706
APA StyleLo Giudice, M., Borgione, E., Giuliano, M., Santa Paola, S., Di Blasi, F. D., Pettinato, R., Romano, C., & Scuderi, C. (2025). Expansion of the Genotypic and Phenotypic Spectrum of TCTN3-Related Joubert Syndrome. Genes, 16(6), 706. https://doi.org/10.3390/genes16060706