
Diversity and Role of Prophages in Pseudomonas aeruginosa: Resistance Genes and Bacterial Interactions


 , , and
, , and 
Abstract

1. Introduction
2. Materials and Methods
2.1. P. aeruginosa Genome
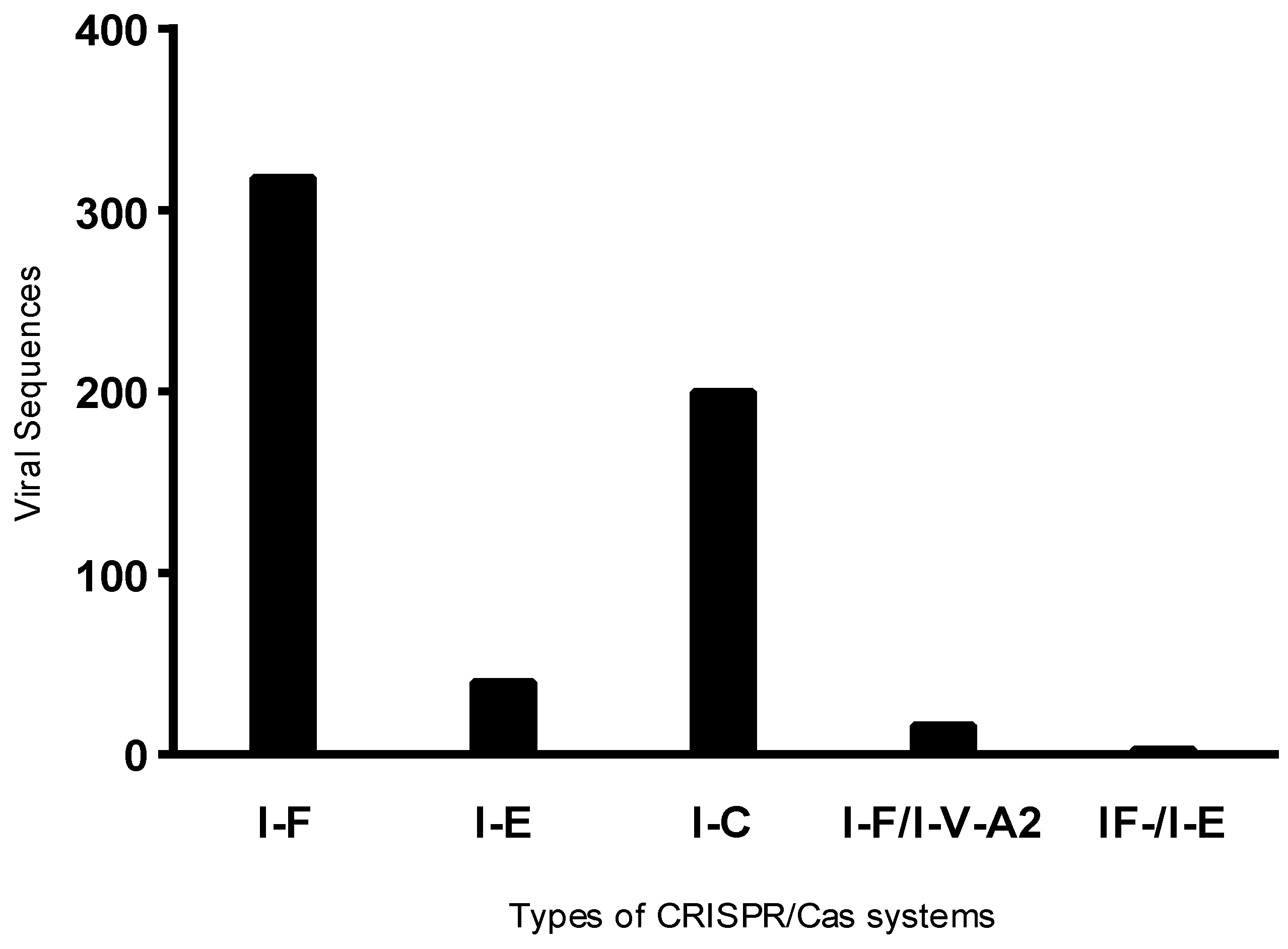
2.2. CRISPR System Identification
2.3. Prophage Identification
2.4. Prophage Distribution in P. aeruginosa Genomes
2.5. Clustering, Phylogeny, and Dendogram
2.6. Identification of Resistance Genes and Virulence Factors in Prophages
2.7. Prophage Life Cycle
2.8. Statistical Correlations
3. Results
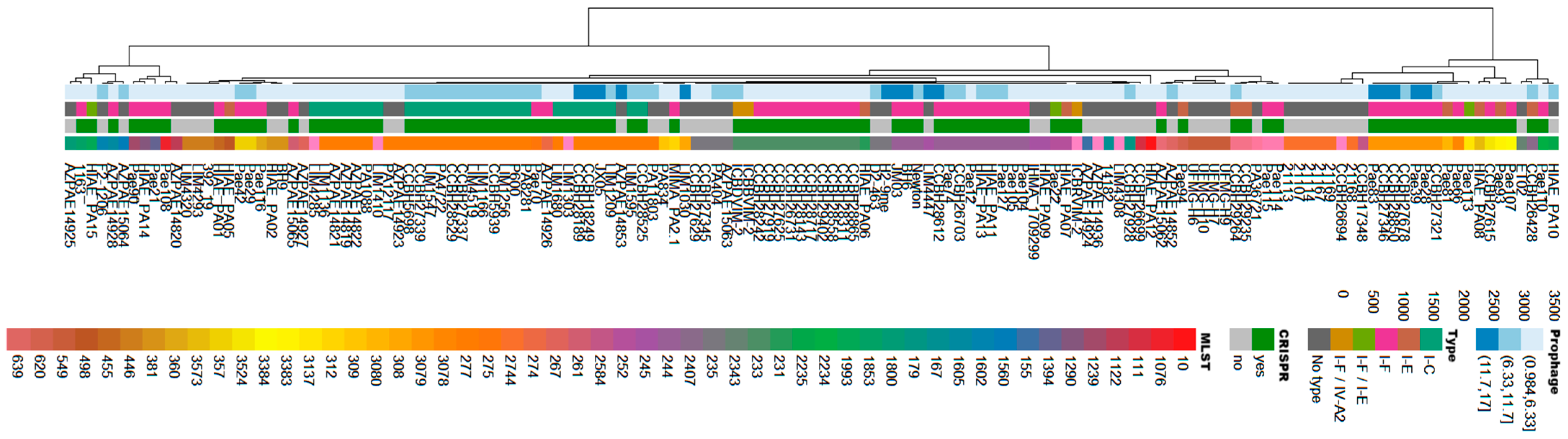
3.1. In Silico Analysis Identifies Prophages in Brazilian Clinical Isolates of P. aeruginosa
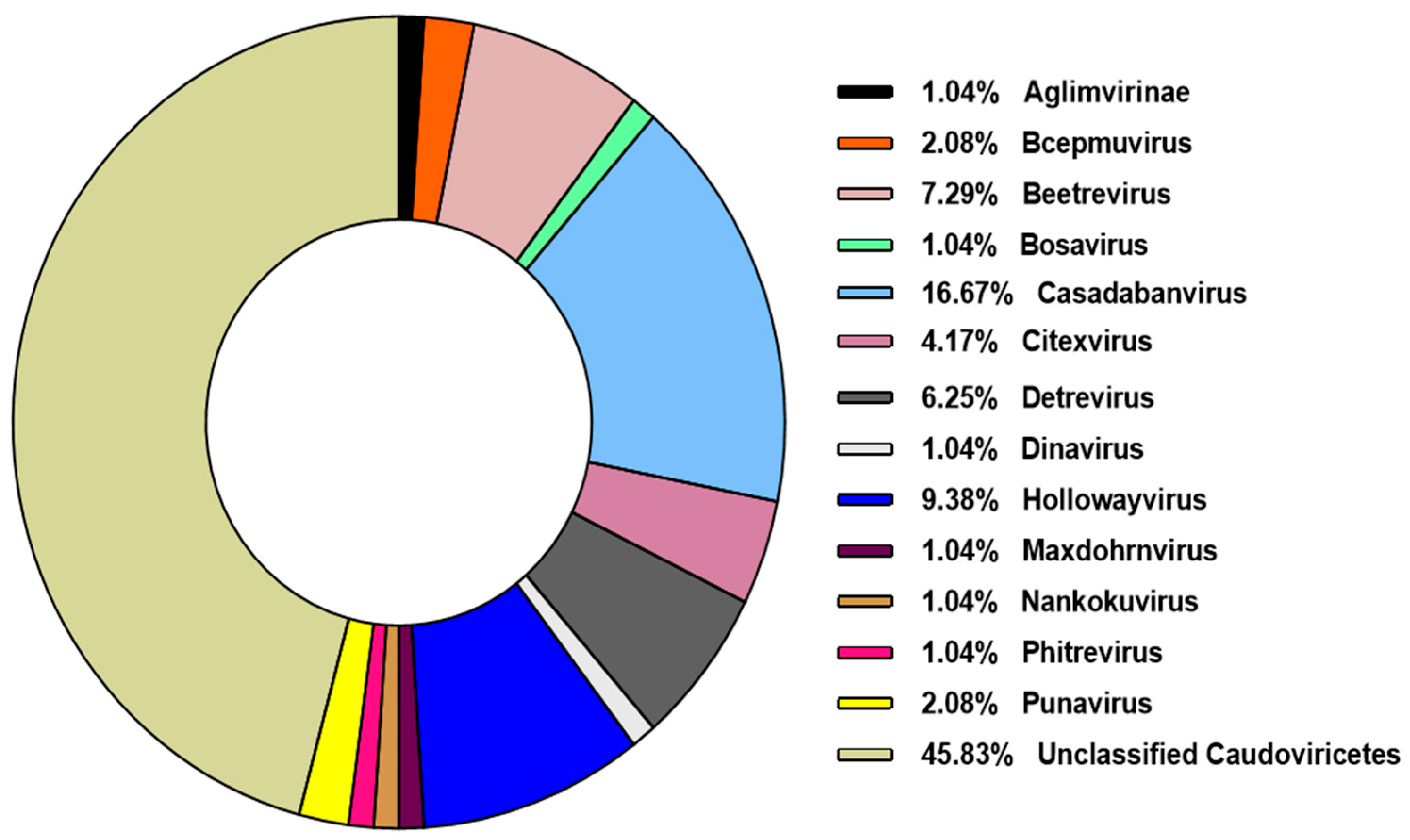
3.2. Caudoviricetes Is the Most Commonly Identified Class of Prophages in P. aeruginosa Genomes
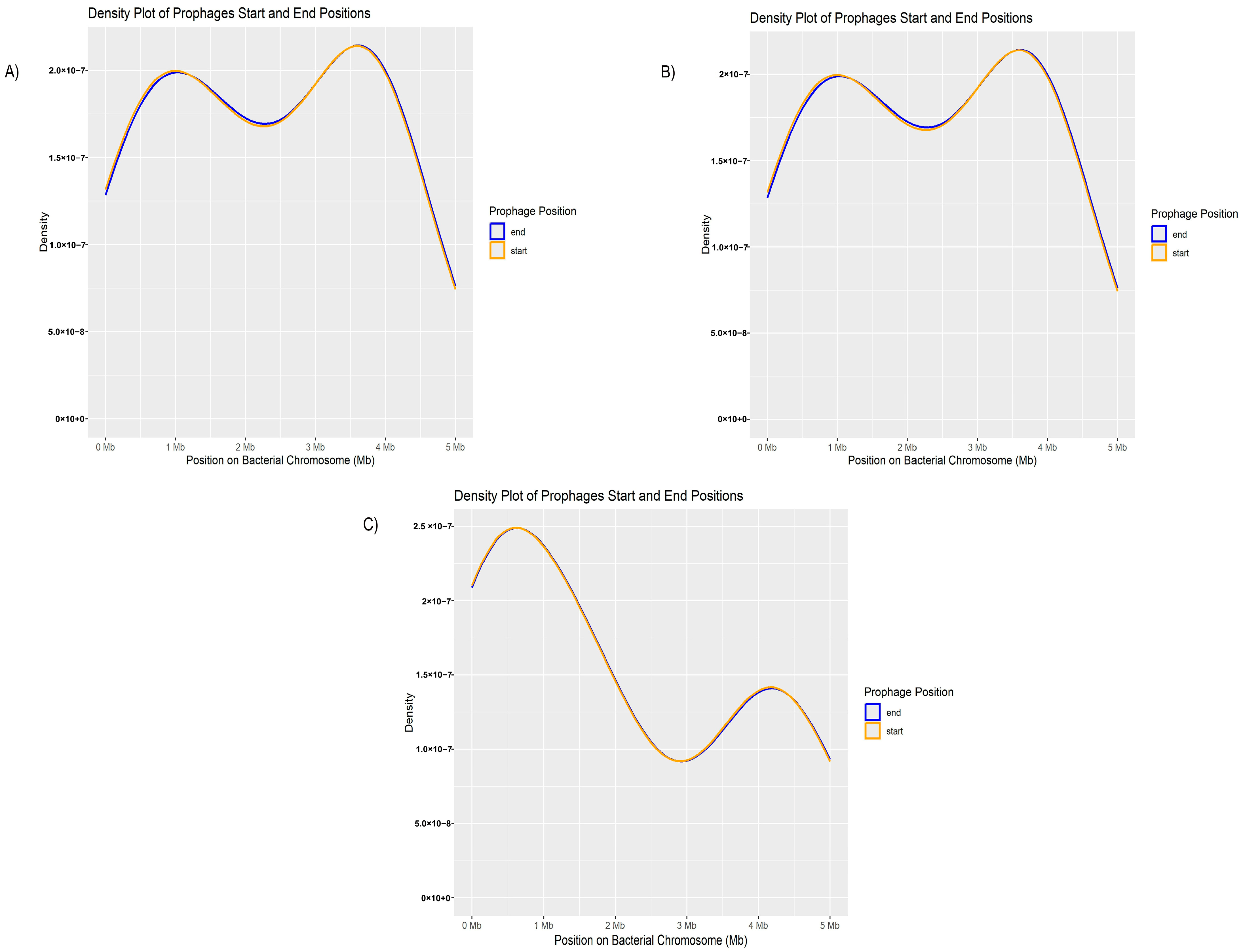
3.3. P. aeruginosa Prophages Do Not Exhibit Unique Insertion Sites
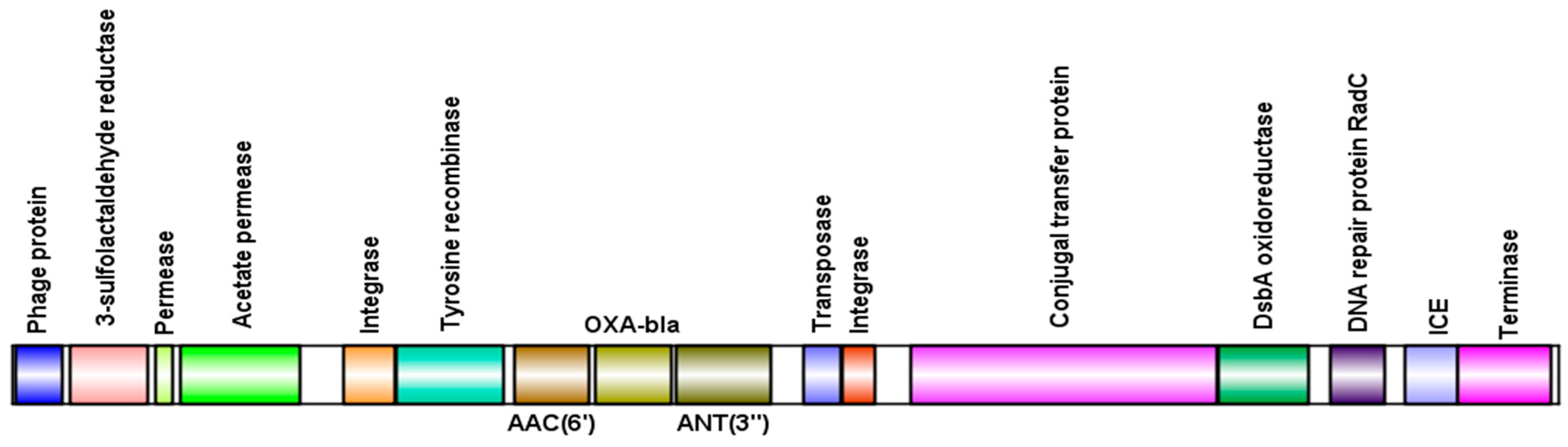
3.4. Presence of Antibiotic Resistance Genes in Viral Sequences
3.5. The Lytic Cycle Is More Frequent in Clinical Isolates of P. aeruginosa
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CRISPR/CAS | Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated genes |
| DNA | Deoxyribonucleic acid |
| ICEs | Integrative and Conjugative Elements |
| PAGIs | Pseudomonas aeruginosa Genomic Islands |
References
- Codjoe, F.; Donkor, E. Carbapenem Resistance: A Review. Med. Sci. 2017, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Bacterial Priority Pathogens List 2024: Bacterial Pathogens of Public Health Importance, to Guide Research, Development, and Strategies to Prevent and Control Antimicrobial Resistance, 1st ed.; World Health Organization: Geneva, Switzerland, 2024. [Google Scholar]
- Qin, S.; Xiao, W.; Zhou, C.; Pu, Q.; Deng, X.; Lan, L.; Liang, H.; Song, X.; Wu, M. Pseudomonas aeruginosa: Pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Signal Transduct. Target. Ther. 2022, 7, 199. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.; Salabarria, A.-C.; Roach, D.R. Phage Therapy in the Resistance Era: Where Do We Stand and Where Are We Going? Clin. Ther. 2020, 42, 1659–1680. [Google Scholar] [CrossRef]
- Davies, E.V.; Winstanley, C.; Fothergill, J.L.; James, C.E. The role of temperate bacteriophages in bacterial infection. FEMS Microbiol. Lett. 2016, 363, fnw015. [Google Scholar] [CrossRef]
- Chevallereau, A.; Pons, B.J.; Van Houte, S.; Westra, E.R. Interactions between bacterial and phage communities in natural environments. Nat. Rev. Microbiol. 2022, 20, 49–62. [Google Scholar] [CrossRef]
- Tsao, Y.-F.; Taylor, V.L.; Kala, S.; Bondy-Denomy, J.; Khan, A.N.; Bona, D.; Cattoir, V.; Lory, S.; Davidson, A.R.; Maxwell, K.L. Phage Morons Play an Important Role in Pseudomonas aeruginosa Phenotypes. J. Bacteriol. 2018, 20. [Google Scholar] [CrossRef] [PubMed]
- Bucher, M.J.; Czyż, D.M. Phage against the Machine: The SIE-ence of Superinfection Exclusion. Viruses 2024, 16, 1348. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.; Casjens, S.R.; Millard, A.D.; Harrison, C.; Gannon, L.; Chattaway, M.A. Genomic analysis of Anderson typing phages of Salmonella Typhimrium: Towards understanding the basis of bacteria-phage interaction. Sci. Rep. 2023, 13, 10484. [Google Scholar] [CrossRef]
- Luz, A.C.D.O.; Da Silva, J.M.A.; Rezende, A.M.; De Barros, M.P.S.; Leal-Balbino, T.C. Analysis of direct repeats and spacers of CRISPR/Cas systems type I-F in Brazilian clinical strains of Pseudomonas aeruginosa. Mol. Genet. Genom. 2019, 294, 1095–1105. [Google Scholar] [CrossRef]
- Xavier, K.V.M.; De Oliveira Luz, A.C.; Silva-Junior, J.W.; De Melo, B.S.T.; De Aragão Batista, M.V.; De Albuquerque Silva, A.M.; De Queiroz Balbino, V.; Leal-Balbino, T.C. Molecular epidemiological study of Pseudomonas aeruginosa strains isolated from hospitals in Brazil by MLST and CRISPR/Cas system analysis. Mol. Genet. Genom. 2025, 300, 33. [Google Scholar] [CrossRef] [PubMed]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef]
- Angiuoli, S.V.; Salzberg, S.L. Mugsy: Fast multiple alignment of closely related whole genomes. Bioinformatics 2011, 27, 334–342. [Google Scholar] [CrossRef]
- Gauthier, C.H.; Cresawn, S.G.; Hatfull, G.F. PhaMMseqs: A new pipeline for constructing phage gene phamilies using MMseqs2. G3 Genes|Genomes|Genet. 2022, 12, jkac233. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, C.H.; Hatfull, G.F. PhamClust: A phage genome clustering tool using proteomic equivalence. mSystems 2023, 8, e00443-23. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program: Table 1. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Farris, J.S. Estimating Phylogenetic Trees from Distance Matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Göker, M.; García-Blázquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular Taxonomy of Phytopathogenic Fungi: A Case Study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef]
- McNair, K.; Bailey, B.A.; Edwards, R.A. PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics 2012, 28, 614–618. [Google Scholar] [CrossRef]
- Turner, D.; Shkoporov, A.N.; Lood, C.; Millard, A.D.; Dutilh, B.E.; Alfenas-Zerbini, P.; Van Zyl, L.J.; Aziz, R.K.; Oksanen, H.M.; Poranen, M.M.; et al. Abolishment of morphology-based taxa and change to binomial species names: 2022 taxonomy update of the ICTV bacterial viruses subcommittee. Arch. Virol. 2023, 168, 74. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://ictv.global/ (accessed on 27 May 2025).
- Pachori, P.; Gothalwal, R.; Gandhi, P. Emergence of antibiotic resistance Pseudomonas aeruginosa in intensive care unit; a critical review. Genes Dis. 2019, 6, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Van Belkum, A.; Soriaga, L.B.; LaFave, M.C.; Akella, S.; Veyrieras, J.-B.; Barbu, E.M.; Shortridge, D.; Blanc, B.; Hannum, G.; Zambardi, G.; et al. Phylogenetic Distribution of CRISPR-Cas Systems in Antibiotic-Resistant Pseudomonas aeruginosa. mBio 2015, 6, e01796-15. [Google Scholar] [CrossRef]
- Galetti, R.; Andrade, L.N.; Varani, A.M.; Darini, A.L.C. SPM-1-producing Pseudomonas aeruginosa ST277 carries a chromosomal pack of acquired resistance genes: An example of high-risk clone associated with ‘intrinsic resistome’. J. Glob. Antimicrob. Resist. 2019, 16, 183–186. [Google Scholar] [CrossRef]
- Argov, T.; Azulay, G.; Pasechnek, A.; Stadnyuk, O.; Ran-Sapir, S.; Borovok, I.; Sigal, N.; Herskovits, A.A. Temperate bacteriophages as regulators of host behavior. Curr. Opin. Microbiol. 2017, 38, 81–87. [Google Scholar] [CrossRef]
- Olszak, T.; Latka, A.; Roszniowski, B.; Valvano, M.A.; Drulis-Kawa, Z. Phage Life Cycles Behind Bacterial Biodiversity. Curr. Med. Chem. 2017, 24, 3987–4001. [Google Scholar] [CrossRef]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef]
- Shah, S.; Das, R.; Chavan, B.; Bajpai, U.; Hanif, S.; Ahmed, S. Beyond antibiotics: Phage-encoded lysins against Gram-negative pathogens. Front. Microbiol. 2023, 14, 1170418. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xie, L.; Yang, M.; Liu, M.; Li, Q.; Wang, P.; Fan, J.; Jin, J.; Luo, C. Synergistic Antibacterial Effect of Phage pB3074 in Combination with Antibiotics Targeting Cell Wall against Multidrug-Resistant Acinetobacter baumannii In Vitro and Ex Vivo. Microbiol. Spectr. 2023, 11, e00341-23. [Google Scholar] [CrossRef]
- Loh, B.; Chen, J.; Manohar, P.; Yu, Y.; Hua, X.; Leptihn, S. A Biological Inventory of Prophages in A. baumannii Genomes Reveal Distinct Distributions in Classes, Length, and Genomic Positions. Front. Microbiol. 2020, 11, 579802. [Google Scholar] [CrossRef] [PubMed]
- Ramisetty, B.C.M.; Sudhakari, P.A. Bacterial ‘Grounded’ Prophages: Hotspots for Genetic Renovation and Innovation. Front. Genet. 2019, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Piel, D.; Bruto, M.; Labreuche, Y.; Blanquart, F.; Goudenège, D.; Barcia-Cruz, R.; Chenivesse, S.; Le Panse, S.; James, A.; Dubert, J.; et al. Phage–host coevolution in natural populations. Nat. Microbiol. 2022, 7, 1075–1086. [Google Scholar] [CrossRef]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 2019, 25, 285–299.e8. [Google Scholar] [CrossRef]
- Naser, I.B.; Hoque, M.M.; Abdullah, A.; Bari, S.M.N.; Ghosh, A.N.; Faruque, S.M. Environmental bacteriophages active on biofilms and planktonic forms of toxigenic Vibrio cholerae: Potential relevance in cholera epidemiology. PLoS ONE 2017, 12, e0180838. [Google Scholar] [CrossRef]
- Balcázar, J.L. Implications of bacteriophages on the acquisition and spread of antibiotic resistance in the environment. Int. Microbiol. 2020, 23, 475–479. [Google Scholar] [CrossRef]
- Haaber, J.; Leisner, J.J.; Cohn, M.T.; Catalan-Moreno, A.; Nielsen, J.B.; Westh, H.; Penadés, J.R.; Ingmer, H. Bacterial viruses enable their host to acquire antibiotic resistance genes from neighbouring cells. Nat. Commun. 2016, 7, 13333. [Google Scholar] [CrossRef]
- Hilbert, M.; Csadek, I.; Auer, U.; Hilbert, F. Antimicrobial resistance-transducing bacteriophages isolated from surfaces of equine surgery clinics—A pilot study. Eur. J. Microbiol. Immunol. 2017, 7, 296–302. [Google Scholar] [CrossRef]
- Da Silva, G.; Domingues, S. Insights on the Horizontal Gene Transfer of Carbapenemase Determinants in the Opportunistic Pathogen Acinetobacter baumannii. Microorganisms 2016, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Silveira, M.C.; Albano, R.M.; Asensi, M.D.; Carvalho-Assef, A.P.D. Description of genomic islands associated to the multidrug-resistant Pseudomonas aeruginosa clone ST277. Infect. Genet. Evol. 2016, 42, 60–65. [Google Scholar] [CrossRef] [PubMed]
- do Nascimento, A.P.B.; Medeiros Filho, F.; Pauer, H.; Antunes, L.C.M.; Sousa, H.; Senger, H.; Albano, R.M.; Trindade Dos Santos, M.; Carvalho-Assef, A.P.D.; Da Silva, F.A.B. Characterization of a SPM-1 metallo-β-lactamase-producing Pseudomonas aeruginosa by comparative genomics and phenotypic analysis. Sci. Rep. 2020, 10, 13192. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Most Common Prophages | Less Common Prophages | ||
|---|---|---|---|
| Phage | Total | Phage | Total |
| Bacteriophage sp. | 147 | Pseudomonas phage PAJU2 | 1 |
| Caudoviricetes sp. | 136 | Escherichia phage P1 | 1 |
| Pseudomonas phage phi3 | 50 | Pseudomonas phage JBD26 | 1 |
| Pseudomonas phage phi297 | 35 | Pseudomonas phage PA8 | 1 |
| Pseudomonas phage H71 | 32 | Pseudomonas phage JBD5 | 1 |
| Pseudomonas phage Dobby | 25 | Burkholderia phage phiE255 | 1 |
| Pseudomonas phage AUS531phi | 24 | Pseudomonas phage vB_Pae_CF125a | 1 |
| Pseudomonas phage phiCTX | 22 | Ralstonia phage Dina | 1 |
| Pseudomonas phage vB_Pae_CF54a | 16 | Pseudomonas phage D3112 | 1 |
| Pseudomonas phage UMP151 | 15 | Pseudomonas phage MP42 | 1 |
| Group Description | ||||||
|---|---|---|---|---|---|---|
| Group | N | Mean | Median | Standard Deviation | Standard Error | |
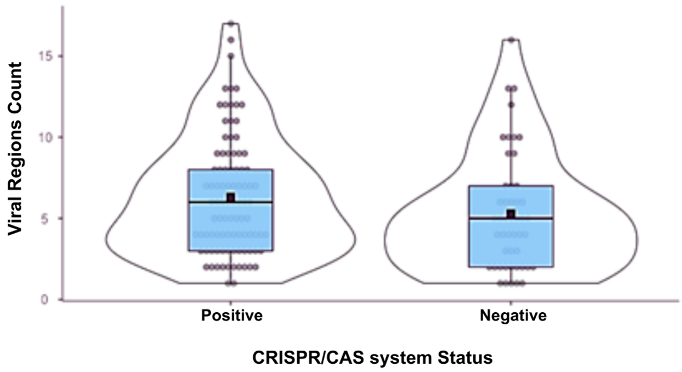
| Phage Region Count | Positive | 93 | 6.31 | 6 | 3.64 | 0.377 |
| Negative | 48 | 5.29 | 5 | 3.64 | 0.525 | |
 | ||||||
| Correlation matrix | ||||||
| Viral region count | ||||||
| CRISPR/Cas status | Spearman’s rho | 0.150 | ||||
| p-value | 0.076 | |||||
| Highest Number of Prophages Identified | Fewest Number of Prophages Identified | ||
|---|---|---|---|
| Isolate | Nº Prophages | Isolate | Nº Prophages |
| CCBH28612 | 17 | AZPAE15065 | 1 |
| H2-9me | 16 | ET02 | 1 |
| Pae28 | 16 | Pae113 | 1 |
| Pae39 | 15 | UFMG-H6 | 1 |
| AZPAE14853 | 13 | UFMG-H7 | 1 |
| CCBH27346 | 13 | UFMG-H9 | 1 |
| JM03 | 13 | UFMG-H10 | 1 |
| LIM1030 | 13 | Pae93 | 2 |
| Pae83 | 13 | Pae94 | 2 |
| BH6 | 12 | Pae110 | 2 |
| Isolate | Start Position | Stop Position | Phage Lenght | CheckV | Resistence Gene |
|---|---|---|---|---|---|
| CCBH28529 | 5516 | 6358 | 28,935 | Low-quality | sul1 |
| CCBH28529 | 55,961 | 56,704 | 69,697 | Low-quality | rmtD |
| CCBH28850 | 7614 | 8111 | 58,067 | Low-quality | dfrA21 |
| CCBH28850 | 8639 | 9478 | 58,067 | Low-quality | sul1 |
| JX05 | 4041 | 5216 | 62,157 | Low-quality | sul1 |
| JX05 | 6351 | 7190 | 62,157 | Low-quality | sul1 |
| JX05 | 9396 | 10,277 | 62,157 | Low-quality | cmx |
| JX05 | 12,157 | 12,954 | 62,157 | Low-quality | aadA7 |
| JX05 | 13,016 | 13,816 | 62,157 | Low-quality | oxa-56 |
| LIM1030 | 2050 | 2889 | 41,494 | Low-quality | sul1 |
| LIM1030 | 2050 | 2889 | 41,494 | Low-quality | sul1 |
| LIM1166 | 7547 | 8722 | 29,997 | Low-quality | cmx |
| LIM1166 | 7547 | 8722 | 29,997 | Low-quality | cmx |
| LIM1410 | 49,583 | 50,215 | 178,563 | Low-quality | oxa-56 |
| LIM1410 | 50,296 | 51,096 | 178,563 | Low-quality | aac(6′)-Ib9 |
| LIM1410 | 51,158 | 51,955 | 178,563 | Low-quality | aadA7 |
| LIM1547 | 1263 | 2174 | 9701 | Low-quality | sul1 |
| LIM1547 | 5140 | 6315 | 9701 | Low-quality | cmx |
| LIM4519 | 7397 | 8572 | 12,669 | Low-quality | cmx |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xavier, K.V.M.; Silva, A.M.d.A.; Luz, A.C.d.O.; da Silva, F.S.C.; de Melo, B.S.T.; Pitta, J.L.d.L.P.; Leal-Balbino, T.C. Diversity and Role of Prophages in Pseudomonas aeruginosa: Resistance Genes and Bacterial Interactions. Genes 2025, 16, 656. https://doi.org/10.3390/genes16060656
Xavier KVM, Silva AMdA, Luz ACdO, da Silva FSC, de Melo BST, Pitta JLdLP, Leal-Balbino TC. Diversity and Role of Prophages in Pseudomonas aeruginosa: Resistance Genes and Bacterial Interactions. Genes. 2025; 16(6):656. https://doi.org/10.3390/genes16060656
Chicago/Turabian StyleXavier, Keyla Vitória Marques, Adrianne Maria de Albuquerque Silva, Ana Carolina de Oliveira Luz, Felipe Santana Caboclo da Silva, Beatriz Souza Toscano de Melo, João Luiz de Lemos Padilha Pitta, and Tereza Cristina Leal-Balbino. 2025. "Diversity and Role of Prophages in Pseudomonas aeruginosa: Resistance Genes and Bacterial Interactions" Genes 16, no. 6: 656. https://doi.org/10.3390/genes16060656
APA StyleXavier, K. V. M., Silva, A. M. d. A., Luz, A. C. d. O., da Silva, F. S. C., de Melo, B. S. T., Pitta, J. L. d. L. P., & Leal-Balbino, T. C. (2025). Diversity and Role of Prophages in Pseudomonas aeruginosa: Resistance Genes and Bacterial Interactions. Genes, 16(6), 656. https://doi.org/10.3390/genes16060656