
Active Protein Network Analysis Reveals Coordinated Modules and Critical Proteins Involving Extracellular Electron Transfer Process


Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Interaction Network
2.2. Gene Expression Data
2.3. Active Protein Network
2.4. Enrichment Analysis
2.5. Visualization
3. Results and Discussion
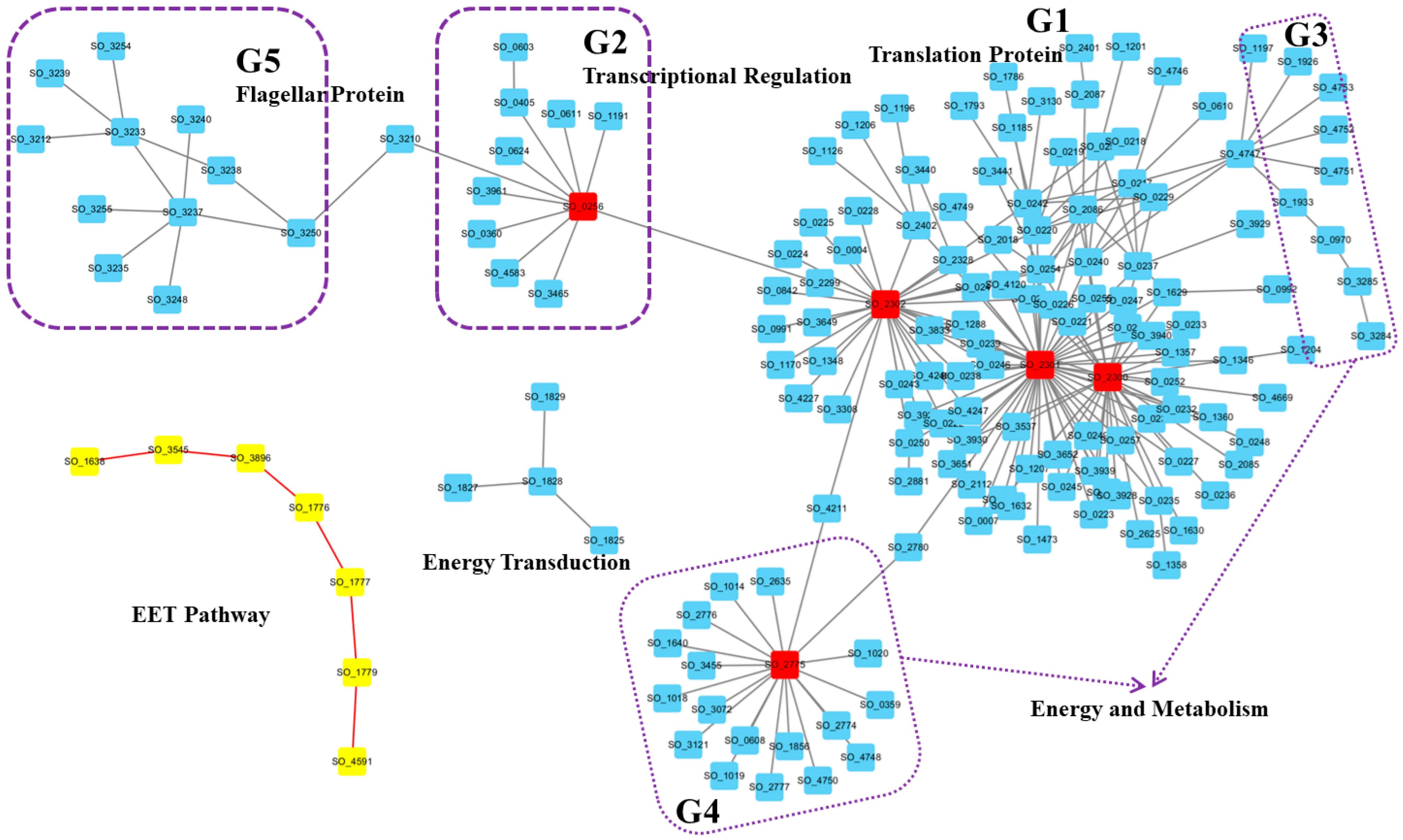
3.1. Highest Activity Networks Were Consistent Under EET Conditions
3.2. Condition-Specific Active Networks Revealed Highly Coordinated Modules Under EET Conditions
3.3. Time-Course Active Networks Analysis Revealed Critical Proteins Driving Cellular Translation Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lovley, D.R.; Holmes, D.E. Electromicrobiology: The ecophysiology of phylogenetically diverse electroactive microorganisms. Nat. Rev. Microbiol. 2022, 20, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Dong, H.; Reguera, G.; Beyenal, H.; Lu, A.; Liu, J.; Yu, H.Q.; Fredrickson, J.K. Extracellular electron transfer mechanisms between microorganisms and minerals. Nat. Rev. Microbiol. 2016, 14, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Logan, B.E.; Rossi, R.; Ragab, A.; Saikaly, P.E. Electroactive microorganisms in bioelectrochemical systems. Nat. Rev. Microbiol. 2019, 17, 307–319. [Google Scholar] [CrossRef]
- Butler, J.E.; Young, N.D.; Lovley, D.R. Evolution of electron transfer out of the cell: Comparative genomics of six Geobacter genomes. BMC Genom. 2010, 11, 40. [Google Scholar] [CrossRef]
- Light, S.H.; Meheust, R.; Ferrell, J.L.; Cho, J.; Deng, D.; Agostoni, M.; Iavarone, A.T.; Banfield, J.F.; D’Orazio, S.E.F.; Portnoy, D.A. Extracellular electron transfer powers flavinylated extracellular reductases in Gram-positive bacteria. Proc. Natl. Acad. Sci. USA 2019, 116, 26892–26899. [Google Scholar] [CrossRef]
- Ding, D.; Shu, C.; Sun, X. Transcriptional regulatory module analysis reveals that bridge proteins reconcile multiple signals in extracellular electron transfer pathways. Proteins 2020, 88, 196–205. [Google Scholar] [CrossRef]
- Zhang, H.; Tang, X.; Munske, G.R.; Zakharova, N.; Yang, L.; Zheng, C.; Wolff, M.A.; Tolic, N.; Anderson, G.A.; Shi, L.; et al. In vivo identification of the outer membrane protein OmcA-MtrC interaction network in Shewanella oneidensis MR-1 cells using novel hydrophobic chemical cross-linkers. J. Proteome Res. 2008, 7, 1712–1720. [Google Scholar] [CrossRef]
- Sturm, G.; Richter, K.; Doetsch, A.; Heide, H.; Louro, R.O.; Gescher, J. A dynamic periplasmic electron transfer network enables respiratory flexibility beyond a thermodynamic regulatory regime. ISME J. 2015, 9, 1802–1811. [Google Scholar] [CrossRef]
- Ding, D.W.; Li, L.; Shu, C.J.; Sun, X. K-shell analysis reveals distinct functional parts in an electron transfer network and its implications for extracellular electron transfer. Front. Microbiol. 2016, 7, 530. [Google Scholar] [CrossRef]
- Zhou, S.; Song, D.; Gu, J.D.; Yang, Y.; Xu, M. Perspectives on microbial electron Ttransfer networks for environmental biotechnology. Front. Microbiol. 2022, 13, 845796. [Google Scholar] [CrossRef]
- Ding, D.W.; Sun, X. A comparative study of network motifs in the integrated transcriptional regulation and protein interaction networks of Shewanella. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, C.J.; Monk, J.; Yang, L.; Ebrahim, A.; Palsson, B.O. Computation of condition-dependent proteome allocation reveals variability in the macro and micro nutrient requirements for growth. PLoS Comput. Biol. 2021, 17, e1007817. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ma, L.; Huo, Y.X. Reconstructing the transcription regulatory network to optimize resource allocation for robust biosynthesis. Trends Biotechnol. 2022, 40, 735–751. [Google Scholar] [CrossRef] [PubMed]
- Li, D.B.; Li, J.; Liu, D.F.; Ma, X.; Cheng, L.; Li, W.W.; Qian, C.; Mu, Y.; Yu, H.Q. Potential regulates metabolism and extracellular respiration of electroactive Geobacter biofilm. Biotechnol. Bioeng. 2019, 116, 961–971. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Taylor, R.C.; Webb Robertson, B.J.; Markillie, L.M.; Serres, M.H.; Linggi, B.E.; Aldrich, J.T.; Hill, E.A.; Romine, M.F.; Lipton, M.S.; Wiley, H.S. Changes in translational efficiency is a dominant regulatory mechanism in the environmental response of bacteria. Integr. Biol. 2013, 5, 1393–1406. [Google Scholar] [CrossRef]
- Barchinger, S.E.; Pirbadian, S.; Sambles, C.; Baker, C.S.; Leung, K.M.; Burroughs, N.J.; El-Naggar, M.Y.; Golbeck, J.H. Regulation of gene expression in Shewanella oneidensis MR-1 during electron acceptor limitation and bacterial nanowire formation. Appl. Environ. Microbiol. 2016, 82, 5428–5443. [Google Scholar] [CrossRef]
- Bar-Joseph, Z.; Gitter, A.; Simon, I. Studying and modelling dynamic biological processes using time-series gene expression data. Nat. Rev. Genet. 2012, 13, 552–564. [Google Scholar] [CrossRef]
- Sambaturu, N.; Pusadkar, V.; Hannenhalli, S.; Chandra, N. PathExt: A general framework for path-based mining of omics-integrated biological networks. Bioinformatics 2021, 37, 1254–1262. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hagberg, A.; Schult, D.; Swart, P. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science conference (SciPy 2008), Pasadena, CA, USA, 19–24 August 2008; Varoquaux, G., Vaught, T., Millman, J., Eds.; pp. 11–15. [Google Scholar]
- Ding, D.W.; Sun, X. Relating translation efficiency to protein networks provides evolutionary insights in Shewanella and its implications for extracellular electron transfer. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 605–613. [Google Scholar] [CrossRef]
- Coursolle, D.; Gralnick, J.A. Modularity of the Mtr respiratory pathway of Shewanella oneidensis strain MR-1. Mol. Microbiol. 2010, 77, 995–1008. [Google Scholar] [CrossRef]
- Maier, T.M.; Myers, C.R. The outer membrane protein Omp35 affects the reduction of Fe(III), nitrate, and fumarate by Shewanella oneidensis MR-1. BMC Microbiol. 2004, 4, 23. [Google Scholar] [CrossRef]
- Elias, D.A.; Monroe, M.E.; Marshall, M.J.; Romine, M.F.; Belieav, A.S.; Fredrickson, J.K.; Anderson, G.A.; Smith, R.D.; Lipton, M.S. Global detection and characterization of hypothetical proteins in Shewanella oneidensis MR-1 using LC-MS based proteomics. Proteomics 2005, 5, 3120–3130. [Google Scholar] [CrossRef]
- Romine, M.F.; Carlson, T.S.; Norbeck, A.D.; McCue, L.A.; Lipton, M.S. Identification of mobile elements and pseudogenes in the Shewanella oneidensis MR-1 genome. Appl. Environ. Microbiol. 2008, 74, 3257–3265. [Google Scholar] [CrossRef]
- Gao, H.; Wang, X.; Yang, Z.K.; Chen, J.; Liang, Y.; Chen, H.; Palzkill, T.; Zhou, J. Physiological roles of ArcA, Crp, and EtrA and their interactive control on aerobic and anaerobic respiration in Shewanella oneidensis. PLoS ONE 2010, 5, e15295. [Google Scholar] [CrossRef]
- Lou, J.; Cai, J.; Hu, X.; Liang, Y.; Sun, Y.; Zhu, Y.; Meng, Q.; Zhu, T.; Gao, H.; Yu, Z.; et al. The stringent starvation protein SspA modulates peptidoglycan synthesis by regulating the expression of peptidoglycan synthases. Mol. Microbiol. 2022, 118, 716–730. [Google Scholar] [CrossRef]
- Kawamoto, J.; Kurihara, T.; Kitagawa, M.; Kato, I.; Esaki, N. Proteomic studies of an Antarctic cold-adapted bacterium, Shewanella livingstonensis Ac10, for global identification of cold-inducible proteins. Extremophiles 2007, 11, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhuo, S.; Jing, X.; Yuan, Y.; Rensing, C.; Zhou, S. Flagella act as Geobacter biofilm scaffolds to stabilize biofilm and facilitate extracellular electron transfer. Biosens. Bioelectron. 2019, 146, 111748. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, Y.; An, J.; Liang, D.; Tian, L.; Zhou, L.; Wang, X.; Li, N. Geobacter autogenically secretes fulvic acid to facilitate the dissimilated iron reduction and vivianite recovery. Environ. Sci. Technol. 2020, 54, 10850–10858. [Google Scholar] [CrossRef]
- Liu, L.; Liu, G.; Zhou, J.; Jin, R. Energy taxis toward redox-active surfaces decreases the transport of electroactive bacteria in saturated porous media. Environ. Sci. Technol. 2021, 55, 5559–5568. [Google Scholar] [CrossRef]
- Balakrishnan, R.; de Silva, R.T.; Hwa, T.; Cremer, J. Suboptimal resource allocation in changing environments constrains response and growth in bacteria. Mol. Syst. Biol. 2021, 17, e10597. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- Gescher, J.S.; Cordova, C.D.; Spormann, A.M. Dissimilatory iron reduction in Escherichia coli: Identification of CymA of Shewanella oneidensis and NapC of E. coli as ferric reductases. Mol. Microbiol. 2008, 68, 706–719. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Function | Number | Expected | Enrichment Fold | FDR | Genes |
|---|---|---|---|---|---|
| translation elongation factor activity (GO:0003746) | 4 | 0.16 | 24.65 | 5.01 × 10−5 | SO_0217, SO_0229, SO_1630, SO_2328 |
| translation initiation factor activity (GO:0003743) | 2 | 0.08 | 24.65 | 1.93 × 10−2 | SO_1204, SO_2300 |
| structural constituent of ribosome (GO:0003735) | 38 | 1.58 | 24.02 | 8.67 × 10−52 | SO_0220, SO_0222, SO_0223, SO_0226, SO_0227, SO_0230, SO_0231, SO_0232, SO_0233, SO_0234, SO_0235, SO_0236, SO_0237, SO_0238, SO_0240, SO_0241, SO_0243, SO_0244, SO_0245, SO_0246, SO_0248, SO_0250, SO_0255, SO_0257, SO_1357, SO_1360, SO_1629, SO_2301, SO_2302, SO_2402, SO_3651, SO_3652, SO_3928, SO_3930, SO_3939, SO_3940, SO_4246, SO_4247 |
| rRNA binding (GO:0019843) | 10 | 0.49 | 20.55 | 2.90 × 10−11 | SO_0220, SO_0227, SO_0238, SO_0241, SO_0247, SO_0255, SO_2112, SO_3537, SO_3928, SO_3930 |
| proton-transporting ATP synthase activity, rotational mechanism (GO:0046933) | 4 | 0.2 | 19.72 | 2.04 × 10−4 | SO_4746, SO_4748, SO_4749, SO_4750 |
| DNA-directed 5′-3′ RNA polymerase activity (GO:0003899) | 2 | 0.12 | 16.44 | 4.58 × 10−2 | SO_0224, SO_0225 |
| protein transmembrane transporter activity (GO:0008320) | 2 | 0.12 | 16.44 | 4.31 × 10−2 | SO_0218, SO_0251 |
| ribosome binding (GO:0043022) | 6 | 0.53 | 11.38 | 9.95 × 10−5 | SO_1170, SO_1346, SO_1632, SO_1793, SO_2300, SO_2625 |
| mRNA binding (GO:0003729) | 4 | 0.41 | 9.86 | 6.04 × 10−3 | SO_0223, SO_0227, SO_2402, SO_3940 |
| NADH dehydrogenase activity (GO:0003954) | 3 | 0.32 | 9.25 | 3.34 × 10−2 | SO_1014, SO_1018, SO_1020 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, D.; Wang, W.; Wang, M.; Xie, J. Active Protein Network Analysis Reveals Coordinated Modules and Critical Proteins Involving Extracellular Electron Transfer Process. Genes 2025, 16, 644. https://doi.org/10.3390/genes16060644
Ding D, Wang W, Wang M, Xie J. Active Protein Network Analysis Reveals Coordinated Modules and Critical Proteins Involving Extracellular Electron Transfer Process. Genes. 2025; 16(6):644. https://doi.org/10.3390/genes16060644
Chicago/Turabian StyleDing, Dewu, Wei Wang, Meineng Wang, and Jianming Xie. 2025. "Active Protein Network Analysis Reveals Coordinated Modules and Critical Proteins Involving Extracellular Electron Transfer Process" Genes 16, no. 6: 644. https://doi.org/10.3390/genes16060644
APA StyleDing, D., Wang, W., Wang, M., & Xie, J. (2025). Active Protein Network Analysis Reveals Coordinated Modules and Critical Proteins Involving Extracellular Electron Transfer Process. Genes, 16(6), 644. https://doi.org/10.3390/genes16060644