
Genetics of Darier’s Disease: New Insights into Pathogenic Mechanisms
 , , , , , ,
, , , , , ,  , , , , ,
, , , , ,  , , , , ,
, , , , ,  , and
, and  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Molecular Pathogenesis
2.1. Darier’s Disease Skin Clinical Features
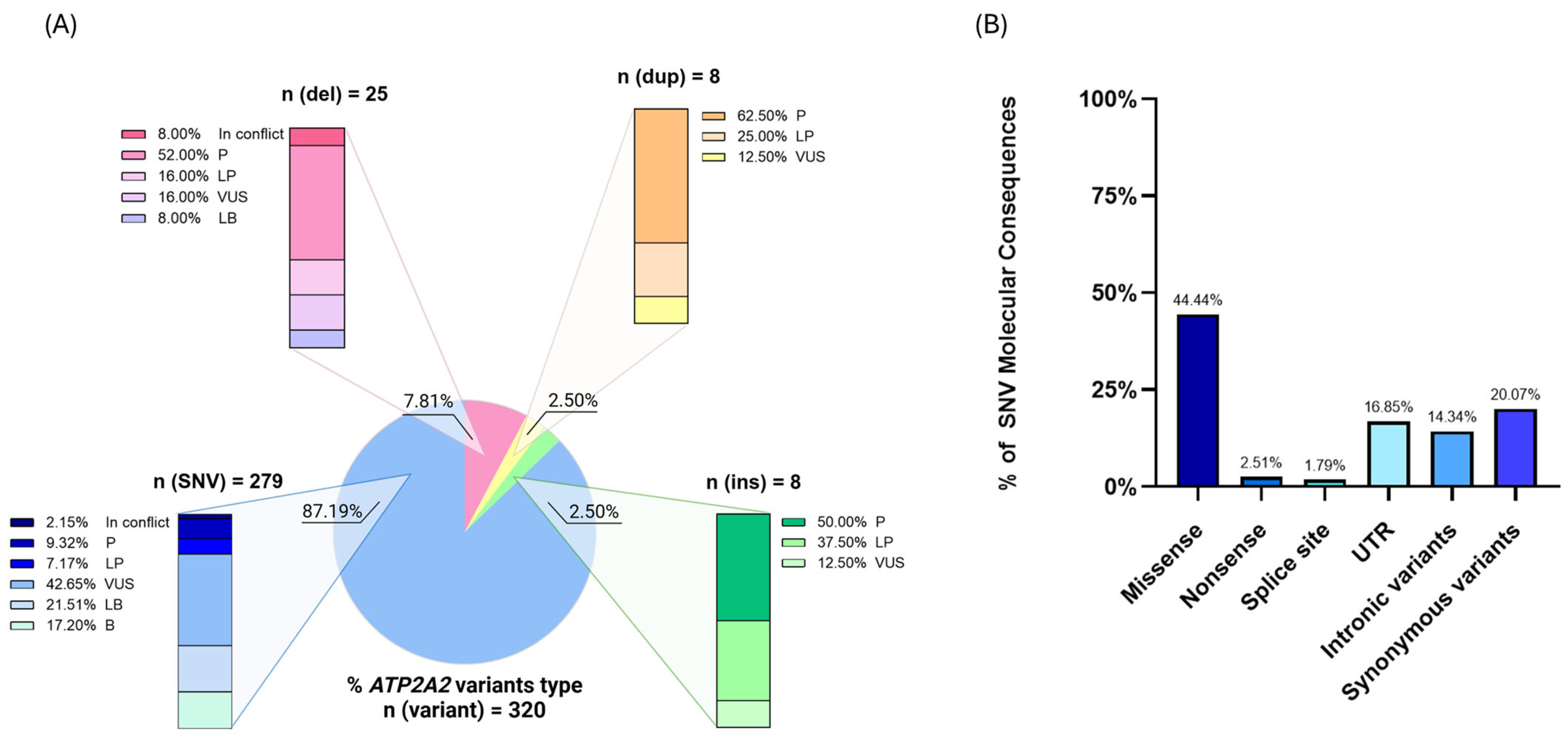
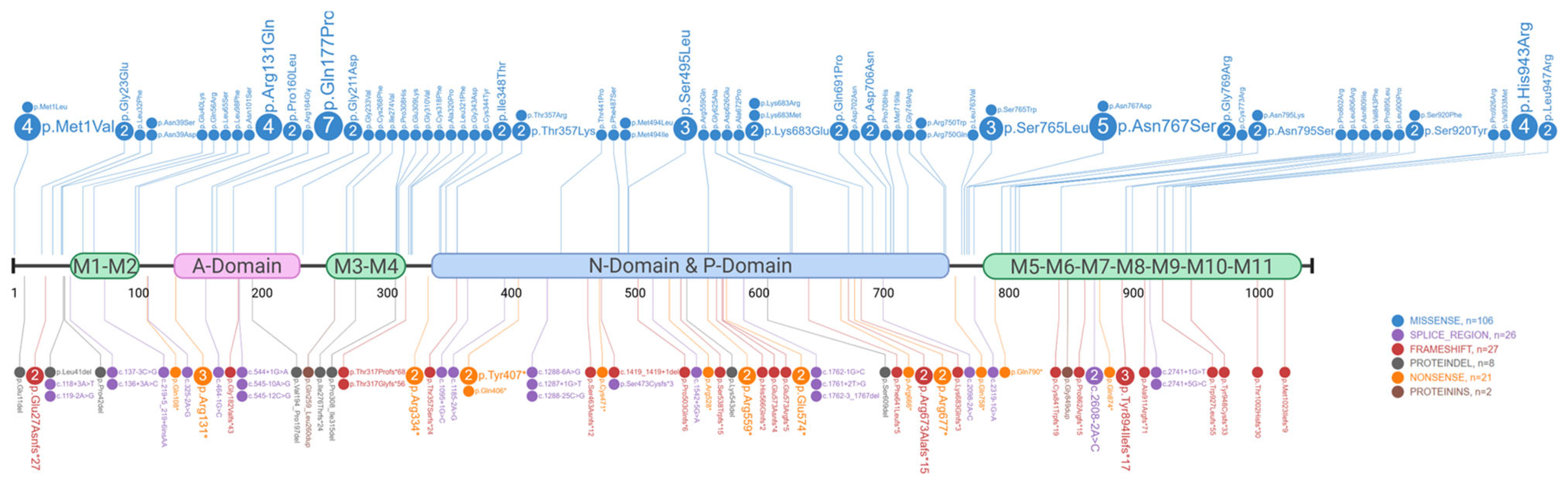
2.2. Genetics of Darier’s Disease
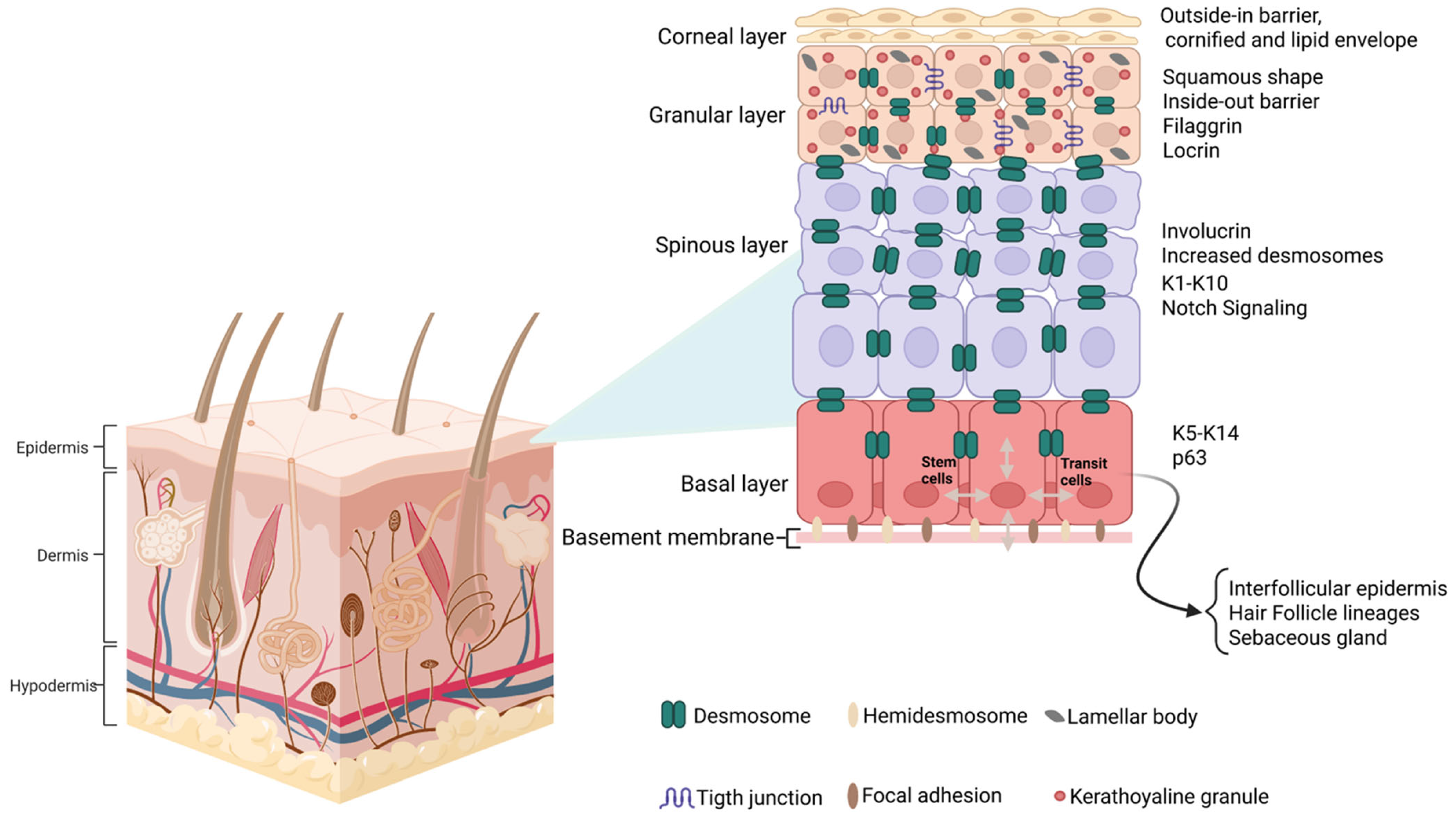
2.3. Cellular Pathophysiology of the Skin Barrier
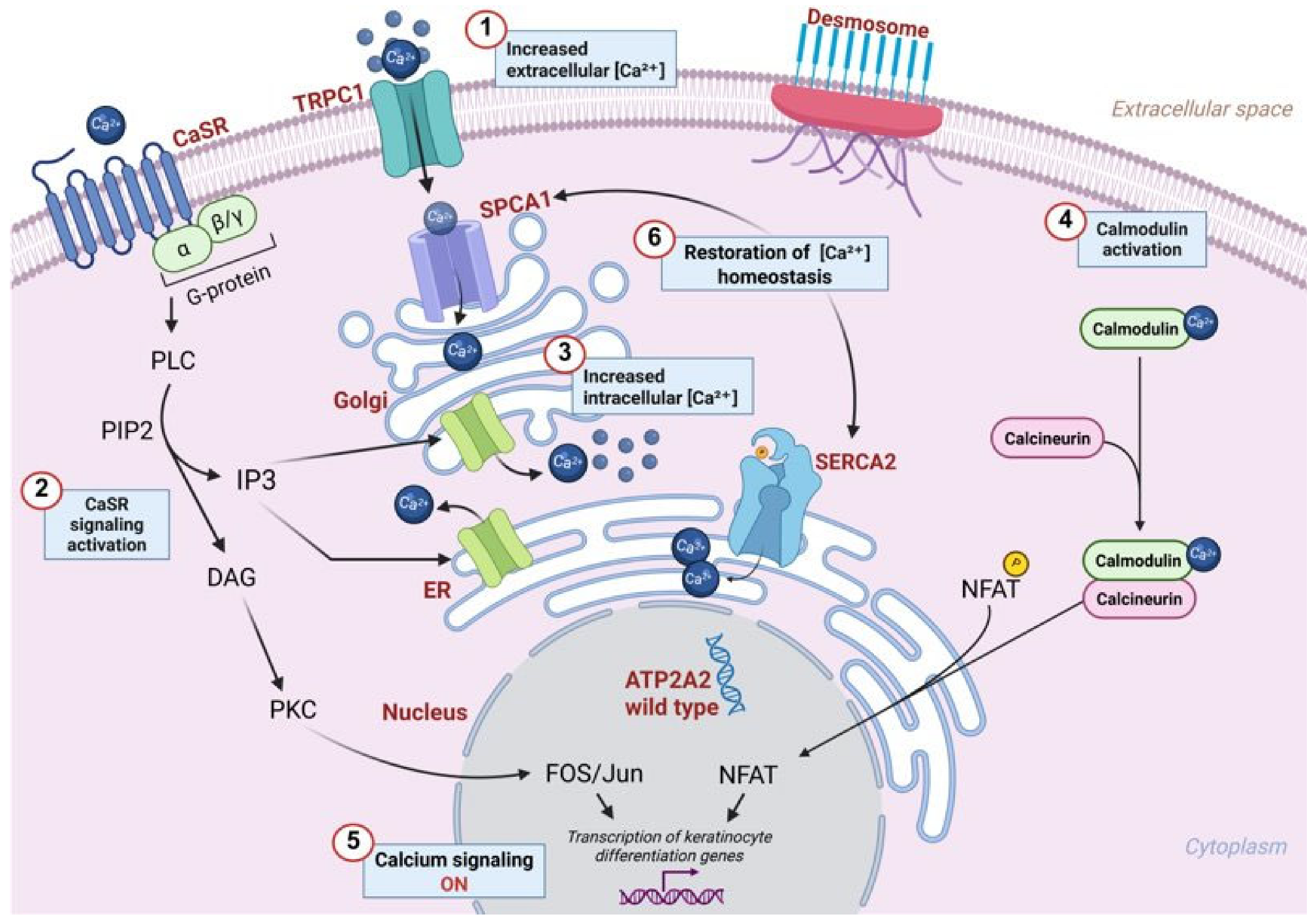
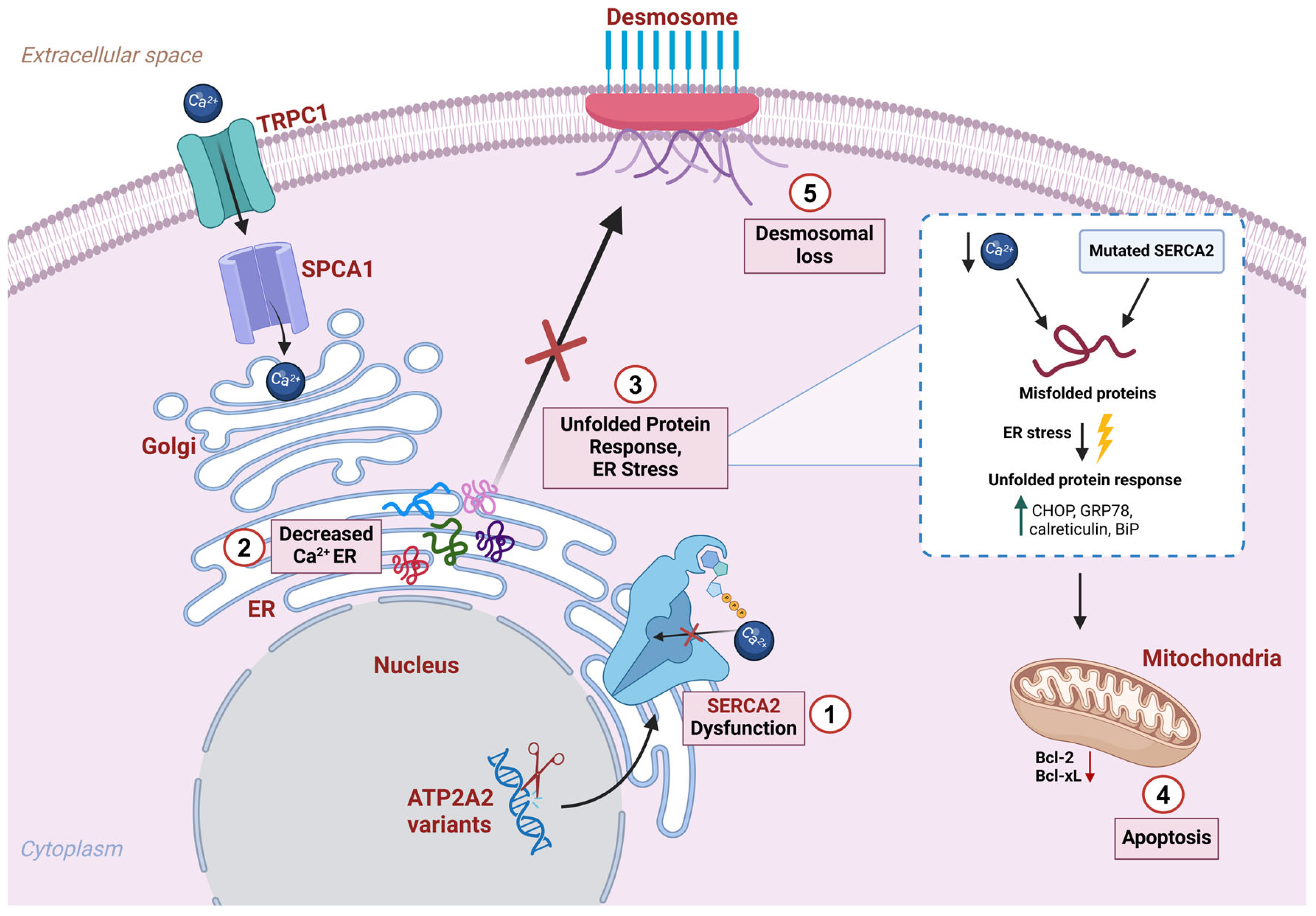
2.4. Skin Barrier Dysfunction Due to SERCA2 Alterations
3. Beyond the Skin: Exploring Systemic Manifestations
4. Treatments
5. Conclusions and Future Perspectives
- Multisystemic nature: DD, an autosomal dominant acantholytic dermatosis, affects skin and mucous membranes, and extends beyond dermatological manifestations to involve neurological, cardiovascular, and metabolic systems. A multi-systemic approach is essential for evaluating patient management and a comprehensive assessment of dermatological, neurological, and cardiac symptoms is crucial. Thereby, a multidisciplinary approach, involving dermatologists, neurologists, cardiologists, and geneticists, is necessary for comprehensive DD management and improved patient outcomes.
- Need for longitudinal studies: Further research, including longitudinal clinical studies and multi-omics analyses, will be crucial to unravel the systemic implications of DD and its comorbidities.
- Unresolved mechanisms: Key gaps remain in understanding the interplay between ER stress, inflammatory pathways, and disease pathogenesis, which could reveal novel therapeutic targets. Moreover, skin microbial imbalances and immune dysfunction studies may open doors to innovative treatments, such as immunomodulatory or microbiome-targeted therapies.
- Symptom-based therapy: The therapeutic approach still relies on retinoids, photodynamic therapy, and surgical excision for the management of skin manifestations, but more targeted treatments are under study.
- Patient-centered focus: Applying personalized medicine and addressing psychosocial impacts, such as quality of life and mental health, should be integral to DD care, given its chronic and stigmatizing nature.
6. Methods
6.1. Literature Search and Selection
6.2. Inclusion/Exclusion Criteria and Data Extraction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Savignac, M.; Edir, A.; Simon, M.; Hovnanian, A. Darier disease: A disease model of impaired calcium homeostasis in the skin. Biochim. Biophys. Acta. 2011, 1813, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Onozuka, T.; Sawamura, D.; Goto, M.; Yokota, K.; Shimizu, H. Possible role of endoplasmic reticulum stress in the pathogenesis of Darier’s disease. J. Dermatol. Sci. 2006, 41, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Takagi, A.; Kamijo, M.; Ikeda, S. Darier disease. J. Dermatol. 2016, 43, 275–279. [Google Scholar] [CrossRef]
- Craddock, N.; Dawson, E.; Burge, S.; Parfitt, L.; Mant, B.; Roberts, Q.; Daniels, J.; Gill, M.; McGuffin, P.; Powell, J.; et al. The gene for Darier’s disease maps to chromosome 12q23–q24. Hum. Mol. Genet. 1993, 12, 1941–1943. [Google Scholar] [CrossRef] [PubMed]
- Hovnanian, A. Darier’s disease: From dyskeratosis to endoplasmic reticulum calcium ATPase deficiency. Biochem. Biophys. Res. Commun. 2004, 322, 1237–1244. [Google Scholar] [CrossRef]
- Shwetha, V.; Sujatha, S.; Yashoda Devi, B.K.; Rakesh, N.; Priyadharshini, R. Spectrum of features in Darier’s disease: A case report with emphasis on differential diagnosis. J. Oral. Biol. Craniofac. Res. 2019, 9, 215–220. [Google Scholar] [CrossRef]
- Aliağaoğlu, C.; Atasoy, M.; Anadolu, R.; Engin, R.I. Comedonal, cornifying and hypertrophic Darier’s disease in the same patient: A Darier combination. J. Dermatol. 2006, 33, 477–480. [Google Scholar] [CrossRef]
- Yeshurun, A.; Ziv, M.; Cohen-Barak, E.; Vered, S.; Rozenman, D.; Sah, M.; Khayat, M.; Polyakov, O.; Amichai, B.; Zlotogorski, A.; et al. An Update on the Cutaneous Manifestations of Darier Disease. J. Cutan. Med. Surg. 2021, 25, 498–503. [Google Scholar] [CrossRef]
- Ettinger, M.; Kimeswenger, S.; Deli, I.; Traxler, J.; Altrichter, S.; Noack, P.; Wikstrom, J.D.; Guenova, E.; Hoetzenecker, W. Darier disease: Current insights and challenges in pathogenesis and management. J. Eur. Acad. Dermatol. Venereol. 2024, 39, 942–951. [Google Scholar] [CrossRef]
- Sakpuwadol, N.; Suchonwanit, P. Papular Acantholytic Dyskeratosis of the Vulva: A Case Report and Literature Review. Clin. Cosmet. Investig. Dermatol. 2025, 18, 31–36. [Google Scholar] [CrossRef]
- Kanakpur, S.; Caculo, D. Rare ocular manifestations in keratosis follicularis (Darier-White disease). Indian J. Ophthalmol. 2017, 65, 874–876. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Adler, E.; Yeshurun, A.; Abayev, L.; Vered, S.; Briscoe, D.; Ziv, M.; Dodiuk-Gad, R.P. Ophthalmic Assessment in Patients With Darier Disease. Am. J. Ophthalmol. 2021, 227, 139–142. [Google Scholar] [CrossRef]
- Burge, S.M.; Wilkinson, J.D. Darier-White disease: A review of the clinical features in 163 patients. J. Am. Acad. Dermatol. 1992, 27, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Dodiuk-Gad, R.; Cohen-Barak, E.; Ziv, M.; Shani-Adir, A.; Amichai, B.; Zlotogorski, A.; Shalev, S. Health-related quality of life among Darier’s disease patients. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 51–56. [Google Scholar] [CrossRef]
- Mourad, A.; Kurwa, H.A.; Haber, R.M. Vesicular Darier’s disease: A case report and review of the English literature of a rare disease variant. SAGE Open Med. Case Rep. 2023, 11, 2050313X231195467. [Google Scholar] [CrossRef]
- Hong, E.; Hu, R.; Posligua, A.; Choate, K.; Durkin, J.R. Acral hemorrhagic Darier disease: A case report of a rare presentation and literature review. JAAD Case Rep. 2023, 31, 93–96. [Google Scholar] [CrossRef]
- Llamas-Velasco, M.; Kempf, W.; Cavelier-Balloy, B.; Camarero-Mulas, C.; Gomez-Vazquez, M.M.; Rütten, A.; Fraga, J.; Fernández-Figueras, M.T. Comedonal Darier’s disease: Six additional cases and a review of this entity. JDDG J. Der Dtsch. Dermatol. Ges. 2020, 18, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Geroldinger-Simic, M.; Jabkowski, J.; Klein, G. Generalized herpes simplex virus infection in Darier’s disease. JDDG J. Der Dtsch. Dermatol. Ges. 2016, 14, 841–842. [Google Scholar] [CrossRef]
- Jacobsen, N.J.O.; Lyons, I.; Hoogendoorn, B.; Burge, S.; Kwok, P.-Y.; O’Donovan, M.C.; Craddock, N.; Owen, M.J. ATP2A2 Mutations in Darier’s Disease and Their Relationship to Neuropsychiatric Phenotypes. Hum. Mol. Genet. 1999, 8, 1631–1636. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Smith, K.; Green, E.; Grozeva, D.; Tavadia, S.; Craddock, N.; Jones, L. Genotype–phenotype correlations in Darier disease: A focus on the neuropsychiatric phenotype. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2018, 177, 717–726. [Google Scholar] [CrossRef]
- Stanisz, H.; Mitteldorf, C.; Janning, H.; Bennemann, A.; Schön, M.P.; Frank, J. Subcellular compartmentalization of STIM1 for the distinction of Darier disease from Hailey-Hailey disease. JDDG J. Ger. Soc. Dermatol. 2022, 20, 1613–1619. [Google Scholar] [CrossRef]
- Amichai, B.; Karpati, M.; Goldman, B.; Peleg, L. Novel mutations in two families with Darier’s disease. Int. J. Dermatol. 2007, 46, 64–67. [Google Scholar] [CrossRef]
- Sakuntabhai, A.; Ruiz-Perez, V.; Carter, S.; Jacobsen, N.; Burge, S.; Monk, S.; Smith, M.; Munro, C.S.; O’Donovan, M.; Craddock, N.; et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat. Genet. 1999, 21, 271–277. [Google Scholar] [CrossRef]
- Sato, K.; Yamasaki, K.; Daiho, T.; Miyauchi, Y.; Takahashi, H.; Ishida-Yamamoto, A.; Nakamura, S.; Iizuka, H.; Suzuki, H. Distinct types of abnormality in kinetic properties of three Darier disease-causing sarco(endo)plasmic reticulum Ca2+-ATPase mutants that exhibit normal expression and high Ca2+ transport activity. J. Biol. Chem. 2004, 279, 35595–35603. [Google Scholar] [CrossRef]
- Ren, Y.-Q.; Gao, M.; Liang, Y.-H.; Hou, Y.-X.; Wang, P.-G.; Sun, L.-D.; Xu, S.-X.; Li, W.; Du, W.-H.; Zhou, F.-S.; et al. Five mutations of ATP2A2 gene in Chinese patients with Darier’s disease and a literature review of 86 cases reported in China. Arch. Dermatol. Res. 2006, 298, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Ringpfeil, F.; Raus, A.; DiGiovanna, J.J.; Korge, B.; Harth, W.; Mazzanti, C.; Uitto, J.; Bale, S.J.; Richard, G. Darier disease—Novel mutations in ATP2A2 and genotype–phenotype correlation. Exp. Dermatol. 2001, 10, 19–27. [Google Scholar] [CrossRef]
- Nellen, R.G.L.; Steijlen, P.M.; van Steensel, M.A.M.; Vreeburg, M.; Frank, J.; van Geel, M. Mendelian Disorders of Cornification Caused by Defects in Intracellular Calcium Pumps: Mutation Update and Database for Variants in ATP2A2 and ATP2C1 Associated with Darier Disease and Hailey–Hailey Disease. Hum. Mutat. 2017, 38, 343–356. [Google Scholar] [CrossRef]
- Frustaci, A.; De Luca, A.; Verardo, R.; Guida, V.; Alfarano, M.; Calvieri, C.; Sansone, L.; Russo, M.A.; Chimenti, C. Novel ATP2A2 Gene Mutation c.118G>A Causing Keratinocyte and Cardiomyocyte Disconnection in Darier Disease. Biomedicines 2024, 12, 1060. [Google Scholar] [CrossRef] [PubMed]
- Tavadia, S.; Tait, R.C.; McDonagh, T.A.; Munro, C.S. Platelet and cardiac function in Darier’s disease. Clin. Exp. Dermatol. 2001, 26, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Leong, I.U.S.; Stuckey, A.; Ahanian, T.; Cederlöf, M.; Wikstrom, J.D. Novel mutations in Darier disease and association to self-reported disease severity. PLoS ONE 2017, 12, e0186356. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Smith, K.; Jones, L.A.; Burge, S.M.; Munro, C.S.; Tavadia, S.; Craddock, N. The neuropsychiatric phenotype in Darier disease. Br. J. Dermatol. 2010, 163, 515–522. [Google Scholar] [CrossRef]
- Amar, Y.; Rogner, D.; Silva, R.L.; Foesel, B.U.; Ud-Dean, M.; Lagkouvardos, I.; Steimle-Grauer, S.A.; Niedermeier, S.; Kublik, S.; Jargosch, M.; et al. Darier’s disease exhibits a unique cutaneous microbial dysbiosis associated with inflammation and body malodour. Microbiome 2023, 11, 162. [Google Scholar] [CrossRef]
- Liu, L.H.; Boivin, G.P.; Prasad, V.; Periasamy, M.; Shull, G.E. Squamous Cell Tumors in Mice Heterozygous for a Null Allele of Atp2a2, Encoding the Sarco(endo)plasmic Reticulum Ca2+-ATPase Isoform 2 Ca2+ Pump. J. Biol. Chem. 2001, 276, 26737–26740. [Google Scholar] [CrossRef]
- Sakuntabhai, A.; Burge, S.; Monk, S.; Hovnanian, A. Spectrum of novel ATP2A2 mutations in patients with Darier’s disease. Hum. Mol. Genet. 1999, 8, 1611–1619. [Google Scholar] [CrossRef]
- Wang, Y.; Bruce, A.T.; Tu, C.; Ma, K.; Zeng, L.; Zheng, P.; Liu, Y. Protein aggregation of SERCA2 mutants associated with Darier disease elicits ER stress and apoptosis in keratinocytes. J. Cell Sci. 2011, 124, 3568–3580. [Google Scholar] [CrossRef]
- Pagliaro, L.; Marchesini, M.; Roti, G. Targeting oncogenic Notch signaling with SERCA inhibitors. J. Hematol. Oncol. 2021, 14, 8. [Google Scholar] [CrossRef]
- Watt, F.M. Stem cell fate and patterning in mammalian epidermis. Curr. Opin. Genet. Dev. 2001, 11, 410–417. [Google Scholar] [CrossRef]
- Moreci, R.S.; Lechler, T. Epidermal structure and differentiation. Curr. Biol. 2020, 30, R144–R149. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Ahn, S.K.; Brown, B.E.; Crumrine, D.; Feingold, K.R. Origin of the Epidermal Calcium Gradient: Regulation by Barrier Status and Role of Active vs Passive Mechanisms. J. Investig. Dermatol. 2002, 119, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D.; Xie, Z.; Tu, C.-L. Calcium regulation of keratinocyte differentiation. Expert. Rev. Endocrinol. Metab. 2012, 7, 461–472. [Google Scholar] [CrossRef]
- Li, Y.; Giovannini, S.; Wang, T.; Fang, J.; Li, P.; Shao, C.; Wang, Y.; Centre, T.; Agostini, M.; Bove, P.; et al. p63: A crucial player in epithelial stemness regulation. Oncogene 2023, 42, 3371–3384. [Google Scholar] [CrossRef]
- Caserta, T.M.; Kommagani, R.; Yuan, Z.; Robbins, D.J.; Mercer, C.A.; Kadakia, M.P. p63 Overexpression Induces the Expression of Sonic Hedgehog. Mol. Cancer Res. 2006, 4, 759–768. [Google Scholar] [CrossRef]
- Nguyen, B.-C.; Lefort, K.; Mandinova, A.; Antonini, D.; Devgan, V.; Della Gatta, G.; Koster, M.I.; Zhang, Z.; Wang, J.; di Vignano, A.T.; et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes. Dev. 2006, 20, 1028–1042. [Google Scholar] [CrossRef]
- Chari, N.S.; A Romano, R.; I Koster, M.; Jaks, V.; Roop, D.; Flores, E.R.; Teglund, S.; Sinha, S.; Gruber, W.; Aberger, F.; et al. Interaction between the TP63 and SHH pathways is an important determinant of epidermal homeostasis. Cell Death Differ. 2013, 20, 1080–1088. [Google Scholar] [CrossRef]
- Abe, Y.; Tanaka, N. Roles of the Hedgehog Signaling Pathway in Epidermal and Hair Follicle Development, Homeostasis, and Cancer. J. Dev. Biol. 2017, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Villanueva, J.; Colome, M.I.; Brisbay, S.; McDonnell, T.J. The Expression and Localization of bcl-2 Protein in Normal Skin and in Non-Melanoma Skin Cancers. Pathol. Res. Pr. 1995, 191, 391–398. [Google Scholar] [CrossRef]
- Lim, X.; Tan, S.H.; Koh, W.L.C.; Chau, R.M.W.; Yan, K.S.; Kuo, C.J.; van Amerongen, R.; Klein, A.M.; Nusse, R. Interfollicular Epidermal Stem Cells Self-Renew via Autocrine Wnt Signaling. Science 2013, 342, 1226–1230. [Google Scholar] [CrossRef]
- Kosumi, H.; Watanabe, M.; Shinkuma, S.; Nohara, T.; Fujimura, Y.; Tsukiyama, T.; Donati, G.; Iwata, H.; Nakamura, H.; Ujiie, H.; et al. Wnt/β-Catenin Signaling Stabilizes Hemidesmosomes in Keratinocytes. J. Investig. Dermatol. 2022, 142, 1576–1586.e2. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.-D.; He, J.; Bazan, H.E. p38 and ERK1/2 Coordinate Cellular Migration and Proliferation in Epithelial Wound Healing. J. Biol. Chem. 2003, 278, 21989–21997. [Google Scholar] [CrossRef] [PubMed]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, 857. [Google Scholar] [CrossRef]
- Prenzel, N.; Fischer, O.M.; Streit, S.; Hart, S.; Ullrich, A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr. Relat. Cancer 2001, 8, 11–31. [Google Scholar] [CrossRef]
- Teng, Y.; Fan, Y.; Ma, J.; Lu, W.; Liu, N.; Chen, Y.; Pan, W.; Tao, X. The PI3K/Akt Pathway: Emerging Roles in Skin Homeostasis and a Group of Non-Malignant Skin Disorders. Cells 2021, 10, 1219. [Google Scholar] [CrossRef]
- Nowell, C.; Radtke, F. Cutaneous Notch Signaling in Health and Disease. Cold Spring Harb. Perspect. Med. 2013, 3, a017772. [Google Scholar] [CrossRef]
- Zaver, S.A.; Sarkar, M.K.; Egolf, S.; Zou, J.; Tiwaa, A.; Capell, B.C.; Gudjonsson, J.E.; Simpson, C.L. Targeting SERCA2 in organotypic epidermis reveals MEK inhibition as a therapeutic strategy for Darier disease. JCI Insight 2023, 8, e170739. [Google Scholar] [CrossRef]
- Bernales, S.; Papa, F.R.; Walter, P. Intracellular Signaling by the Unfolded Protein Response. Annu. Rev. Cell Dev. Biol. 2006, 22, 487–508. [Google Scholar] [CrossRef]
- Scheuner, D.; Kaufman, R.J. The unfolded protein response: A pathway that links insulin demand with β-cell failure and diabetes. Endocr. Rev. 2008, 29, 317–333. [Google Scholar] [CrossRef]
- Caspersen, C.; Pedersen, P.S.; Treiman, M. The Sarco/Endoplasmic Reticulum Calcium-ATPase 2b Is an Endoplasmic Reticulum Stress-inducible Protein. J. Biol. Chem. 2000, 275, 22363–22372. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef]
- Bongiorno, M.R.; Arico, M. The behaviour of Bcl-2, Bax and Bcl-x in Darier’s disease. Br. J. Dermatol. 2002, 147, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Majore, S.; Biolcati, G.; Barboni, L.; Cannistraci, C.; Binni, F.; Crisi, A.; Picardo, M.; Grammatico, P. ATP2C1 gene mutation analysis in Italian patients with Hailey-Hailey disease. J. Investig. Dermatol. 2005, 125, 933–935. [Google Scholar] [CrossRef]
- Engin, B.; Kutlubay, Z.; Erkan, E.; Tüzün, Y. Darier disease: A fold (intertriginous) dermatosis. Clin. Dermatol. 2015, 33, 448–451. [Google Scholar] [CrossRef]
- Behne, M.J.; Tu, C.-L.; Aronchik, I.; Epstein, E.; Bench, G.; Bikle, D.D.; Pozzan, T.; Mauro, T.M. Human Keratinocyte ATP2C1 Localizes to the Golgi and Controls Golgi Ca2+ Stores. J. Investig. Dermatol. 2003, 121, 688–694. [Google Scholar] [CrossRef]
- Behne, M.J.; Tu, C.-L.; Aronchik, I.; Epstein, E.; Bench, G.; Bikle, D.D.; Pozzan, T.; Mauro, T.M. Hailey–Hailey disease and tight junctions: Claudins 1 and 4 are regulated by ATP2C1 gene encoding Ca2+/Mn2+ ATPase SPCA1 in cultured keratinocytes. Exp. Dermatol. 2012, 21, 586–591. [Google Scholar] [CrossRef]
- Micaroni, M.; Perinetti, G.; Berrie, C.P.; Mironov, A.A. The SPCA1 Ca2+ Pump and Intracellular Membrane Trafficking. Traffic 2010, 11, 1315–1333. [Google Scholar] [CrossRef]
- Ambur, A.; Zaidi, A.; Dunn, C.; Nathoo, R. Impaired calcium signalling and neuropsychiatric disorders in Darier disease: An exploratory review. Exp. Dermatol. 2022, 31, 1302–1310. [Google Scholar] [CrossRef]
- Kang, S.-Y.; Lee, S.-Y.; Park, J.-S.; Kim, J.-C.; Chung, B.-Y.; Park, C.-W.; Kim, H.-O. Darier Disease with Psoriasis. Medicina 2022, 58, 902. [Google Scholar] [CrossRef] [PubMed]
- Kassar, S.; Charfeddine, C.; Zribi, H.; Tounsi-Kettiti, H.; Bchetnia, M.; Jerbi, E.; Cassio, D.; Mokni, M.; Abdelhak, S.; Ben Osman, A.; et al. Immunohistological study of involucrin expression in Darier’s disease skin. J. Cutan. Pathol. 2008, 35, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, M.; Burner, T.; Sharma, A.; Chang, Y.-T.; Lackner, A.; Prompsy, P.; Deli, I.M.; Traxler, J.; Wahl, G.; Altrichter, S.; et al. Th17-associated cytokines IL-17 and IL-23 in inflamed skin of Darier disease patients as potential therapeutic targets. Nat. Commun. 2023, 14, 7470. [Google Scholar] [CrossRef]
- Chen, C.-C.; Chen, B.-R.; Wang, Y.; Curman, P.; Beilinson, H.A.; Brecht, R.M.; Liu, C.C.; Farrell, R.J.; de Juan-Sanz, J.; Charbonnier, L.-M.; et al. Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) activity is required for V(D)J recombination. J. Exp. Med. 2021, 218, e20201708. [Google Scholar] [CrossRef] [PubMed]
- Miracco, C.; Pietronudo, F.; Mourmouras, V.; Pellegrino, M.; Onorati, M.; Mastrogiulio, M.G.; Cantarini, L.; Luzi, P. Possible Implication of Local Immune Response in Darier’s Disease: An Immunohistochemical Characterization of Lesional Inflammatory Infiltrate. Mediators Inflamm. 2010, 2010, 350304. [Google Scholar] [CrossRef]
- Reiter, O.; Leshem, A.; Alexander-Shani, R.; Brandwein, M.; Cohen, Y.; Yeshurun, A.; Ziv, M.; Elinav, E.; Hodak, E.; Dodiuk-Gad, R.P. Bacterial Skin Dysbiosis in Darier Disease. Dermatol. 2024, 240, 443–452. [Google Scholar] [CrossRef]
- Yamamoto, T.; Aoyama, Y. Role of pro-inflammatory cytokines in the pathophysiology of herpes simplex virus superinfection in Darier’s disease. J. Dermatol. 2021, 48, 1607–1611. [Google Scholar] [CrossRef]
- Thiagarajan, M.; Sankarasubramanian, A.; Narasimhan, M. Darier disease with oral and esophageal involvement: A case report. Indian J. Dent. Res. 2011, 22, 843–846. [Google Scholar] [CrossRef]
- Klec, C.; Ziomek, G.; Pichler, M.; Malli, R.; Graier, W.F. Calcium signaling in ß-cell physiology and pathology: A revisit. Dec. Int. J. Mol. Sci. 2019, 20, 6110. [Google Scholar] [CrossRef]
- Cederlöf, M.; Curman, P.; Ahanian, T.; Leong, I.U.; Brismar, K.; Bachar-Wikstrom, E.; Wikstrom, J.D. Darier disease is associated with type 1 diabetes: Findings from a population-based cohort study. J. Am. Acad. Dermatol. 2019, 81, 1425–1426. [Google Scholar] [CrossRef]
- Aizawa, T.; Yada, T.; Asanuma, N.; Sato, Y.; Ishihara, F.; Hamakawa, N.; Yaekura, K.; Hashizume, K. Effects of thapsigahgin, an intracellular CA2+ pump inhibitor, on insulin release by rat pancreatic B-cell. Life Sci. 1995, 57, 1375–1381. [Google Scholar] [CrossRef]
- Tersey, S.A.; Nishiki, Y.; Templin, A.T.; Cabrera, S.M.; Stull, N.D.; Colvin, S.C.; Evans-Molina, C.; Rickus, J.L.; Maier, B.; Mirmira, R.G. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 2012, 61, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Marhfour, I.; Lopez, X.M.; Lefkaditis, D.; Salmon, I.; Allagnat, F.; Richardson, S.J.; Morgan, N.G.; Eizirik, D.L. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 2012, 55, 2417–2420. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Ahanian, T.; Curman, P.; Leong, I.U.S.; Brismar, K.; Bachar-Wikstrom, E.; Cederlöf, M.; Wikstrom, J.D. Metabolic phenotype in Darier disease: A cross-sectional clinical study. Diabetol. Metab. Syndr. 2020, 12, 1–4. [Google Scholar] [CrossRef]
- Lian, C.Y.; Zhai, Z.Z.; Li, Z.F.; Wang, L. High fat diet-triggered non-alcoholic fatty liver disease: A review of proposed mechanisms. Chem.-Biol. Interact. 2020, 330, 109199. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef]
- Baba-Aissa, F.; Raeymaekers, L.; Wuytack, F.; Dode, L.; Casteels, R. Distribution and isoform diversity of the organellar Ca2+ pumps in the brain. Mol. Chem. Neuropathol. 1998, 33, 199–208. [Google Scholar] [CrossRef]
- Baba-Aissa, F.; Raeymaekers, L.; Wuytack, F.; De Greef, C.; Missiaen, L.; Casteels, R. Distribution of the organellar Ca2+ transport ATPase SERCA2 isoforms in the cat brain. Brain Res. 1996, 743, 141–153. [Google Scholar] [CrossRef]
- Salvador, J.M.; Berengena, M.; Sepulveda, M.R.; Mata, A.M. Distribution of the Intracellular Ca2+-ATPase Isoform 2b in Pig Brain Subcellular Fractions and Cross-Reaction with a Monoclonal Antibody Raised against the Enzyme Isoform 1. J. Biochem. 2001, 129, 621–626. [Google Scholar] [CrossRef]
- Jameson, C.; Boulton, K.A.; Silove, N.; Nanan, R.; Guastella, A.J. Ectodermal origins of the skin-brain axis: A novel model for the developing brain, inflammation, and neurodevelopmental conditions. Mol. Psychiatry 2023, 28, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kazuno, A.; Nakajima, K.; Kusumi, I.; Tsuboi, T.; Kato, T. Loss of function mutations in ATP2A2 and psychoses: A case report and literature survey. Psychiatry Clin. Neurosci. 2016, 70, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Ishiwata, M.; Weitemier, A.Z.; Shoji, H.; Monai, H.; Miyamoto, H.; Yamakawa, K.; Miyakawa, T.; McHugh, T.J.; Kato, T. Brain-specific heterozygous loss-of-function of ATP2A2, endoplasmic reticulum Ca2+ pump responsible for Darier’s disease, causes behavioral abnormalities and a hyper-dopaminergic state. Hum. Mol. Genet. 2021, 30, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Illes, P.; Verkhratsky, A.; Tang, Y. Pathological ATPergic Signaling in Major Depression and Bipolar Disorder. Front. Mol. Neurosci. 2020, 12, 331. [Google Scholar] [CrossRef]
- Celli, A.; Mackenzie, D.S.; Zhai, Y.; Tu, C.-L.; Bikle, D.D.; Holleran, W.M.; Uchida, Y.; Mauro, T.M. SERCA2-Controlled Ca2+-Dependent Keratinocyte Adhesion and Differentiation Is Mediated via the Sphingolipid Pathway: A Therapeutic Target for Darier’s Disease. J. Investig. Dermatol. 2012, 132, 1188–1195. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, J.; Zhao, K.; Wu, S.; Chen, X.; Hu, W. The Role of Calcium and Iron Homeostasis in Parkinson’s Disease. Brain Sci. 2024, 14, 88. [Google Scholar] [CrossRef]
- Pani, B.; Cornatzer, E.; Cornatzer, W.; Shin, D.-M.; Pittelkow, M.R.; Hovnanian, A.; Ambudkar, I.S.; Singh, B.B. Up-Regulation of Transient Receptor Potential Canonical 1 (TRPC1) following Sarco(endo)plasmic Reticulum Ca2+ ATPase 2 Gene Silencing Promotes Cell Survival: A Potential Role for TRPC1 in Darier’s Disease. Mol. Biol. Cell 2006, 17, 4446–4458. [Google Scholar] [CrossRef]
- Curman, P.; Jebril, W.; Larsson, H.; Bachar-Wikstrom, E.; Cederlöf, M.; Wikstrom, J.D. Darier disease is associated with neurodegenerative disorders and epilepsy. Sci. Rep. 2024, 14, 7109. [Google Scholar] [CrossRef]
- Baker, D.L.; Hashimoto, K.; Grupp, I.L.; Ji, Y.; Reed, T.; Loukianov, E.; Grupp, G.; Bhagwhat, A.; Hoit, B.; Walsh, R.; et al. Targeted Overexpression of the Sarcoplasmic Reticulum Ca 2+ -ATPase Increases Cardiac Contractility in Transgenic Mouse Hearts. Circ. Res. 1998, 83, 1205–1214. [Google Scholar] [CrossRef]
- Del Monte, F.; Harding, S.E.; Schmidt, U.; Matsui, T.; Kang, Z.B.; Dec, G.W.; Gwathmey, J.K.; Rosenzweig, A.; Hajjar, R.J. Restoration of Contractile Function in Isolated Cardiomyocytes From Failing Human Hearts by Gene Transfer of SERCA2a. Circulation 1999, 100, 2308–2311. [Google Scholar] [CrossRef]
- Stüdeli, R.; Jung, S.; Mohacsi, P.; Perruchoud, S.; Castiglioni, P.; Wenaweser, P.; Heimbeck, G.; Feller, M.; Hullin, R. Diastolic Dysfunction in Human Cardiac Allografts is Related with Reduced SERCA2a Gene Expression. Am. J. Transplant. 2006, 6, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Zhang, Z.Y.; Andersson, K.B.; Husberg, C.; Enger, U.H.; Ræder, M.G.; Christensen, G.; Louch, W.E. Cardiomyocyte-specific disruption of Serca2 in adult mice causes sarco(endo)plasmic reticulum stress and apoptosis. Cell Calcium 2011, 49, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Lorenz, J.N.; Lasko, V.M.; Nieman, M.L.; Huang, W.; Wang, Y.; Wieczorek, D.W.; Shull, G.E. SERCA2 Haploinsufficiency in a Mouse Model of Darier Disease Causes a Selective Predisposition to Heart Failure. Biomed. Res. Int. 2015, 2015, 251598. [Google Scholar] [CrossRef] [PubMed]
- Bachar-Wikstrom, E.; Curman, P.; Ahanian, T.; Leong, I.U.S.; Larsson, H.; Cederlöf, M.; Wikstrom, J.D. Darier disease is associated with heart failure: A cross-sectional case-control and population based study. Sci. Rep. 2020, 10, 6886. [Google Scholar] [CrossRef]
- Hanna, N.; Lam, M.; Fleming, P.; Lynde, C.W. Therapeutic Options for the Treatment of Darier’s Disease: A Comprehensive Review of the Literature. J. Cutan. Med. Surg. 2022, 26, 280–290. [Google Scholar] [CrossRef]
- Zamiri, M.; Munro, C.S. Successful treatment with oral alitretinoin in women of childbearing potential with Darier’s disease. Br. J. Dermatol. 2013, 169, 709–710. [Google Scholar] [CrossRef]
- Avery, H.L.; Hughes, B.R.; Coley, C.; Cooper, H.L. Clinical improvement in Darier’s disease with photodynamic therapy. Australas. J. Dermatol. 2010, 51, 32–35. [Google Scholar] [CrossRef]
- Hagino, T.; Nakano, H.; Saeki, H.; Kanda, N. A Case of Darier’s Disease with a Novel Missense Mutation in ATP2A2 Successfully Treated with Calcipotriol/Betamethasone Dipropionate Two-Compound Ointment. Clin. Cosmet. Investig. Dermatol. 2022, 15, 367–372. [Google Scholar] [CrossRef]
- Muto, Y.; Kinjyo, K.; Asano, Y. Successful treatment of Darier’s disease with apremilast and review of reported cases. J. Dermatol. 2025, 52, 138–141. [Google Scholar] [CrossRef]
- Dreyfus, I.; Maza, A.; Rodriguez, L.; Merlos, M.; Texier, H.; Rousseau, V.; Sommet, A.; Mazereeuw-Hautier, J. Botulinum toxin injections as an effective treatment for patients with intertriginous Hailey-Hailey or Darier disease: An open-label 6-month pilot interventional study. Orphanet. J. Rare Dis. 2021, 16, 93. [Google Scholar] [CrossRef]
- Hunt, M.; Wang, N.; Pupinyo, N.; Curman, P.; Torres, M.; Jebril, W.; Chatzinikolaou, M.; Lorent, J.; Silberberg, G.; Bansal, R.; et al. Dantrolene corrects cellular disease features of Darier disease and may be a novel treatment. EMBO Mol. Med. 2024, 16, 1986–2001. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, M.; Nishiyama, C.; Takagi, A.; Nakano, N.; Hara, M.; Ikeda, S.; Okumura, K.; Ogawa, H. Cyclooxygenase-2 inhibition restores ultraviolet B-induced downregulation of ATP2A2/SERCA2 in keratinocytes: Possible therapeutic approach of cyclooxygenase-2 inhibition for treatment of Darier disease. Br. J. Dermatol. 2012, 166, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Jfri, A.; Litvinov, I.V.; Netchiporouk, E. Naltrexone for the Treatment of Darier and Hailey-Hailey Diseases. J. Cutan. Med. Surg. 2019, 23, 453–454. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Involved Amino Acid Position (n = 17) | Patients (n = 62) | Variants (n = 30) | Type | Protein Domain | ACMG Classification (P/LP/VUS) | Severity of DD | Skin Phenotype | Neuropsychiatric Disorder | Cardiac Defects | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| p.Met1 | n = 4 | c.1A>G p.Met1Val | Missense | Start codon | P | Mild/Moderate | Hyperkeratotic patches on abdomen. | Major depressive/Bipolar disorder. | N.a. | [23,30,32] |
| n = 1 | c.1A>T p.Met1Leu | Missense | Start codon | P | N.a. | N.a. | Bipolar disorder. | N.a. | [30] | |
| p.Gly23 | n = 2 | c.68G>A p.Gly23Glu | Missense | S1 | P | N.a. | N.a. | N.a. | N.a. | [26,30] |
| c.118 (splice site) | n = 1 | c.118+3A>T | Splice site | S1 | VUS | Severe | Excoriated papules neck and seborrheic areas trunk, extensive hyperkeratotic flexural patches, and dystrophic toe nails. | - | - | [30] |
| n = 1 | c.118G>A p.Glu40Lys | Splice site | S1 | VUS | Severe | Extensive greasy, crusted, yellow–brown papules on the seborrheic areas. | Depressive syndrome with a failed attempt of suicide. | Palpitations and chest pain at rest. | [31] | |
| p.Arg131 | n = 4 | c.392G>A p.Arg131Gln | Missense | A domain | P | Mild/Moderate | Extensive confluated verrucous plaques in seborrheic areas, V-shaped notches. | N.a. | - | [24,30,32] |
| n = 3 | c.391C>T p.Arg131* | Nonsense | A domain | LP | Moderate/Severe | Hyperkeratotic lesions on the upper trunk and scalp with severe exacerbation during summer. | - | N.a. | [25] | |
| p.Pro160 | n = 2 | c.479C>T p.Pro160Leu | Missense | A domain | LP | Severe | Disseminated hyperkeratotic papules and confluating patches on trunk and extremities. | Learning disability and depressive symptoms. | - | [29,30] |
| p.Gln177 | n = 7 | c.530A>C p.Gln177Pro | Missense | A domain | LP | Moderate | Few skin and oral lesions. Warty, greasy, hyperkeratotic papules and plaques over the scalp, upper trunk, and forearms. Large subcutaneous cysts over his face. Numerous hyperkeratotic papules on the trunk, subungual hyperkeratosis, and few oral lesions. Nail changes and few skin lesions located on the upper chest and flexures. | Depression NOS. | ECG abnormalities (prolonged QT interval). | [25] |
| p.Thr317 | n = 1 | c.948del p.Thr317Profs*68 | Frameshift | S4 | LP | Moderate | N.a. | Psychiatric disorder NOS. Suicide attempt. Epilepsy. | N.a. | [23] |
| n = 1 | c.949_956del p.Thr317Glyfs*56 | Frameshift | S4 | LP | Moderate | N.a. | Major depressive disorder. Suicide attempt. Investigations for a blackout. | N.a. | [23] | |
| p.Ile348 | n = 2 | c.1043T>C p.Ile348Thr | Missense | P domain | P | Moderate/Severe | Recurrent infections and eczema-like perioral lesions. | Depression. | N.a. | [23,29] |
| p.Thr357 | n = 2 | c.1070C>A p.Thr357Lys | Missense | P domain | LP | Moderate | N.a. | Major depressive disorder. Investigations for hearing problems. | N.a. | [23,26] |
| n = 1 | c.1070C>G p.Thr357Arg | Missense | P domain | LP | Mild | N.a. | - | N.a. | [23] | |
| n = 1 | c.1070_1082del p.Thr357Serfs*24 | Frameshift | P domain | LP | Mild | Hyperkeratotic papules in seborrheic areas. | - | - | [30] | |
| p.Ser495 | n = 3 | c.1484C>T p.Ser495Leu | Missense | N domain | LP | Moderate | N.a. | Major depressive disorder. | N.a. | [23,26] |
| p.Arg673 | n = 2 | c.2017del p.Arg673Alafs*15 | Frameshift | P domain | LP | Moderate | N.a. | Major depressive disorder. Investigations for fainting episodes. | N.a. | [23,26] |
| p.Lys683 | n = 2 | c.2047A>G p.Lys683Glu | Missense | P domain | LP | Mild–Moderate | N.a. | N.a. | N.a. | [29,30] |
| n = 1 | c.2046insC p.Lys683Glnfs*3 | Frameshift | P domain | LP | Moderate | N.a. | Anxiety disorder NOS. | N.a. | [23] | |
| n = 1 | c.2048A>T p.Lys683Met | Missense | P domain | LP | Moderate | N.a. | Investigations for blackouts. | N.a. | [23] | |
| n = 1 | c.2048A>G p.Lys683Arg | Missense | P domain | LP | Mild–Moderate | Classical distribution | N.a. | - | [29] | |
| p.Ser765 | n = 3 | c.2294C>T p.Ser765Leu | Missense | M5 | LP | Mild/Moderate | Lower back, knee folds, and waist. | N.a. | [23,33] | |
| n = 1 | c.2294C>G p.Ser765Trp | Missense | M5 | LP | Mild | Hyperkeratotic papules in seborrheic areas. | Dyslexia and mild apraxia. | - | [30] | |
| p.Asn767 | n = 5 | c.2300A>G p.Asn767Ser | Missense | M5 | P | Mild/Moderate | N.a. | Major depressive disorder. Treatment for hearing problems. Investigations for poor memory. | N.a. | [23,32] |
| n = 1 | c.2299A>G p.Asn767Asp | Missense | M5 | P | Mild–Moderate | Eruptions on scalp, neck, trunk, limbs, and nails. | - | N.a. | [28] | |
| p.Asn795 | n = 2 | c.2384A>G p.Asn795Ser | Missense | M6 | VUS | Moderate | Widespread hyperkeratotic papules incl dorsum of hands, longitudinal leukonychia, and palmar pits. | Anxiety disorder NOS. | N.a. | [23,30] |
| n = 1 | c.2385T>G p.Asn795Lys | Missense | M6 | VUS | Severe | Entire body and pharynx. | N.a. | N.a. | [33] | |
| p.Tyr894 | n = 3 | c.2678dup p.Tyr894Ilefs*17 | Frameshift | M7-M8 | LP | Mild | N.a. | Major depressive disorder. | N.a. | [23,32] |
| p.Ser920 | n = 1 | c.2759C>T p.Ser920Phe | Missense | M8–M9 | LP | Moderate | N.a. | Depression NOS. | N.a. | [23] |
| n = 2 | c.2759C>A p.Ser920Tyr | Missense | M8–M9 | LP | Severe | N.a. | Investigations for one-sided weakness. Medical notes report “adjustment reaction” to relapse in DD. | N.a. | [23] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moschella, B.; Busciglio, S.; Ambrosini, E.; Cesarini, S.; Caramanna, L.; Zanelli, S.; Cannizzaro, I.R.; Luberto, A.; Taiani, A.; Treccani, M.; et al. Genetics of Darier’s Disease: New Insights into Pathogenic Mechanisms. Genes 2025, 16, 619. https://doi.org/10.3390/genes16060619
Moschella B, Busciglio S, Ambrosini E, Cesarini S, Caramanna L, Zanelli S, Cannizzaro IR, Luberto A, Taiani A, Treccani M, et al. Genetics of Darier’s Disease: New Insights into Pathogenic Mechanisms. Genes. 2025; 16(6):619. https://doi.org/10.3390/genes16060619
Chicago/Turabian StyleMoschella, Barbara, Sabrina Busciglio, Enrico Ambrosini, Sofia Cesarini, Luca Caramanna, Sara Zanelli, Ilenia Rita Cannizzaro, Anita Luberto, Antonietta Taiani, Mirko Treccani, and et al. 2025. "Genetics of Darier’s Disease: New Insights into Pathogenic Mechanisms" Genes 16, no. 6: 619. https://doi.org/10.3390/genes16060619
APA StyleMoschella, B., Busciglio, S., Ambrosini, E., Cesarini, S., Caramanna, L., Zanelli, S., Cannizzaro, I. R., Luberto, A., Taiani, A., Treccani, M., De Sensi, E., Caggiati, P., Azzoni, C., Bottarelli, L., Lorusso, B., Lagrasta, C. A. M., Montanaro, A., Pagliaro, L., Zamponi, R., ... Percesepe, A. (2025). Genetics of Darier’s Disease: New Insights into Pathogenic Mechanisms. Genes, 16(6), 619. https://doi.org/10.3390/genes16060619