
Olig1/2 Drive Astrocytic Glioblastoma Proliferation Through Transcriptional Co-Regulation of Various Cyclins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
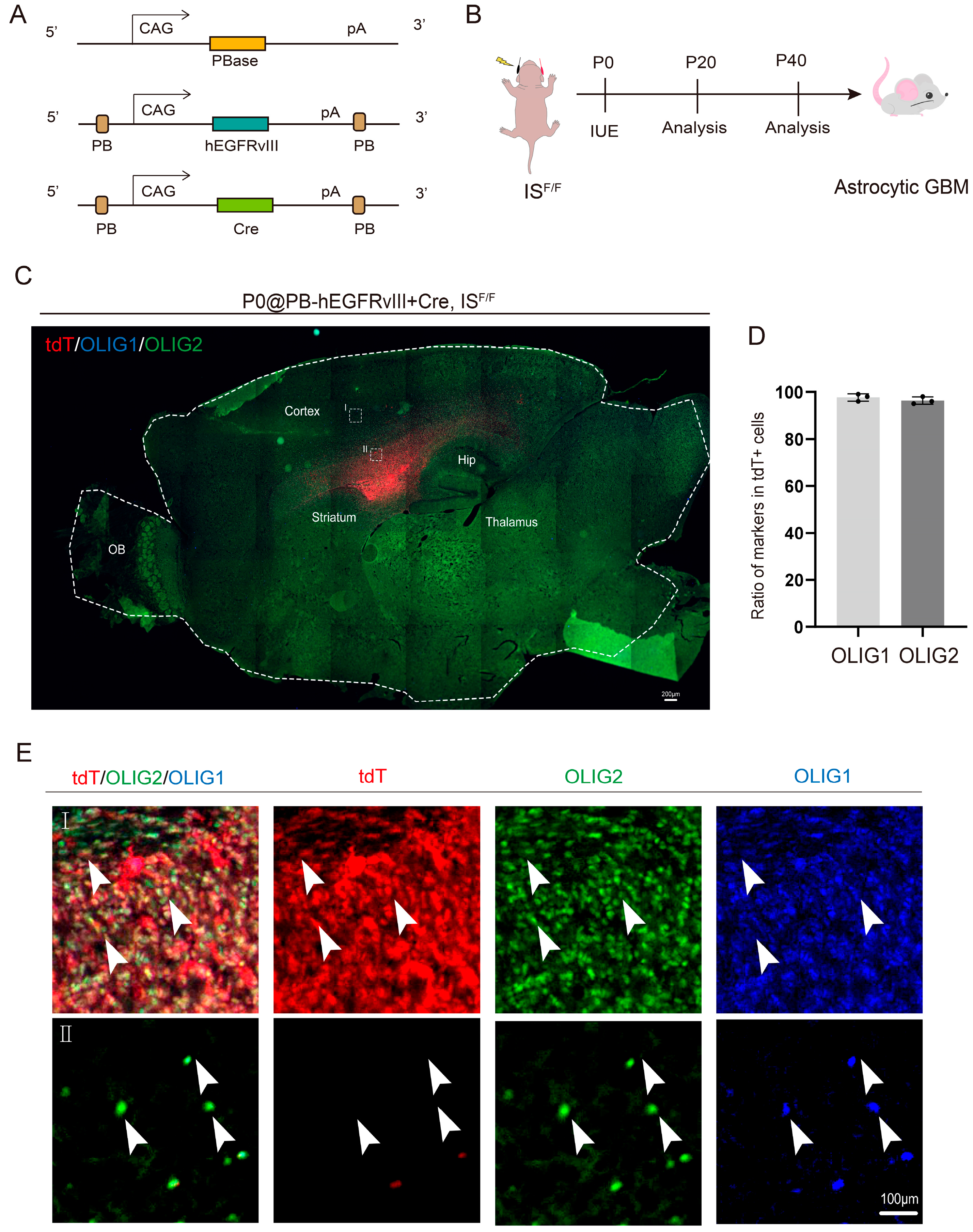
2.2. GBM Mouse Model Generation
2.3. Plasmid Construction
2.4. In Utero Electroporation
2.5. Immunofluorescence Staining
2.6. GEPIA
2.7. UALCAN
2.8. TIMER
2.9. CUT&Tag-Seq
2.10. Image Acquisition and Quantitative Analysis
3. Results
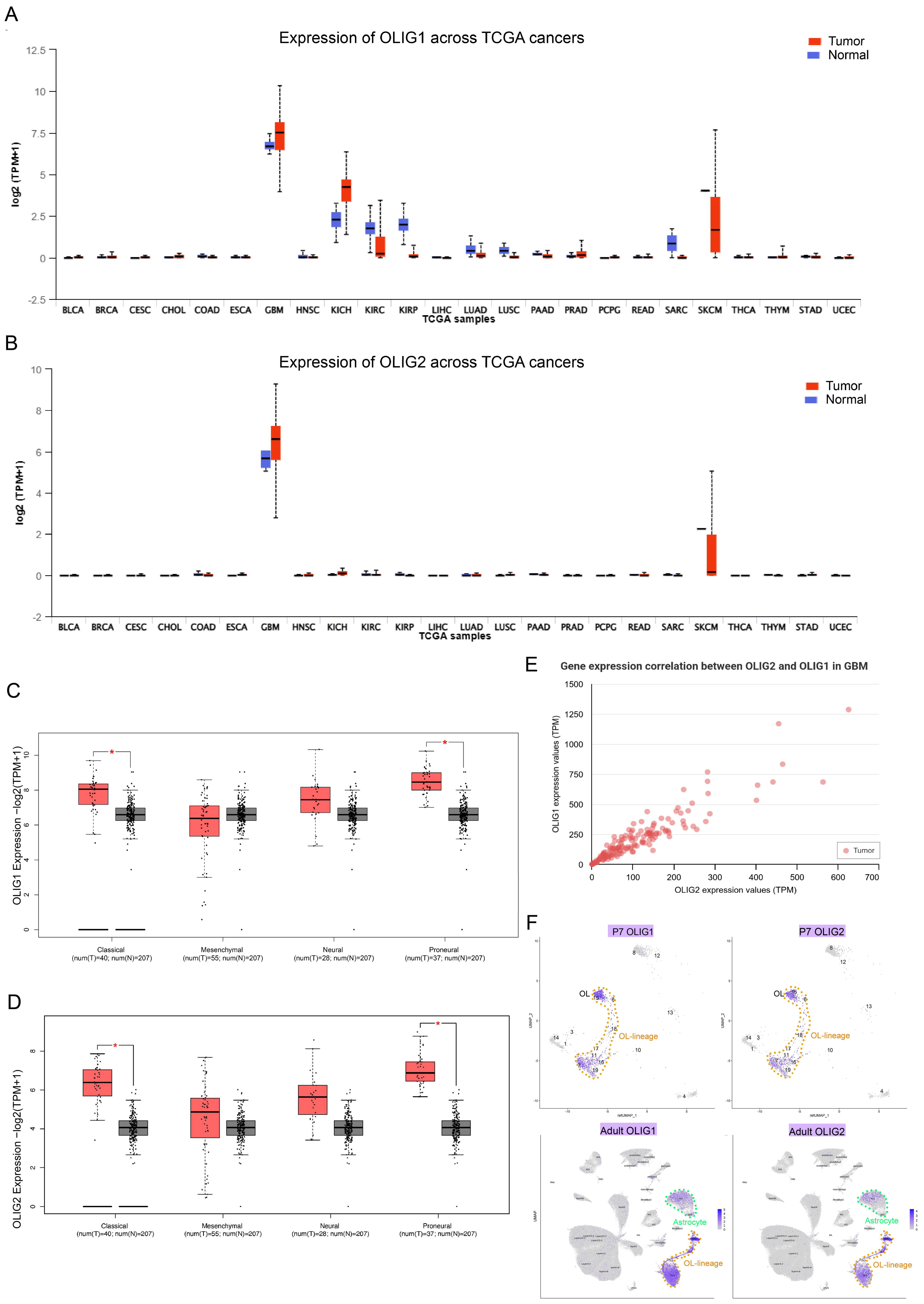
3.1. OLIG1 and OLIG2 Exhibit High Co-Expression in Glioblastoma
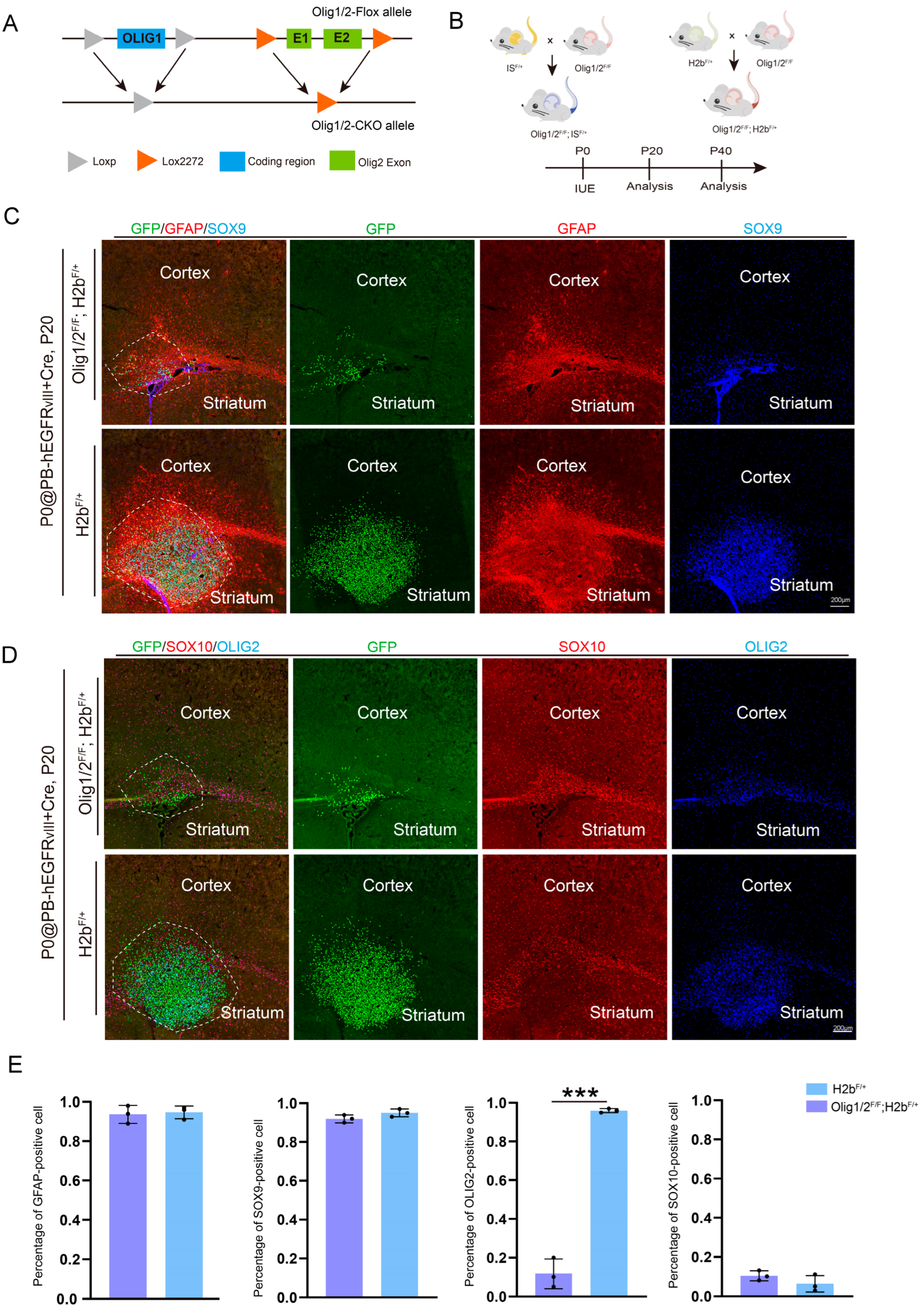
3.2. The Knockout of Olig1 and Olig2 in Astrocytic GBM Does Not Affect Astrocytic Characteristics
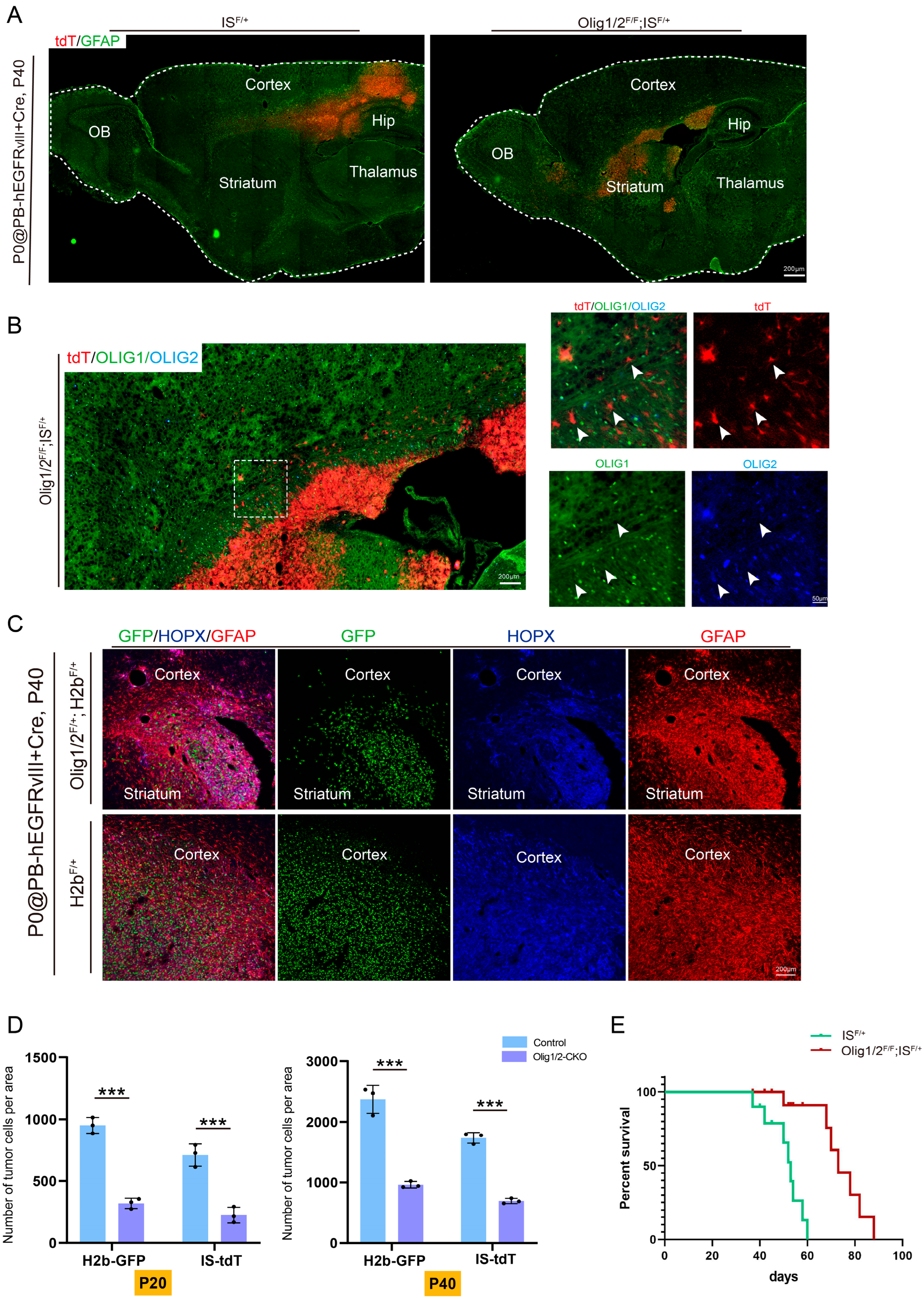
3.3. Olig1/2 Knockout Reduced Tumor Growth Without Affecting Invasion
3.4. Olig1/2 Reduced Tumor Growth by Inhibiting Glioma Cell Proliferation
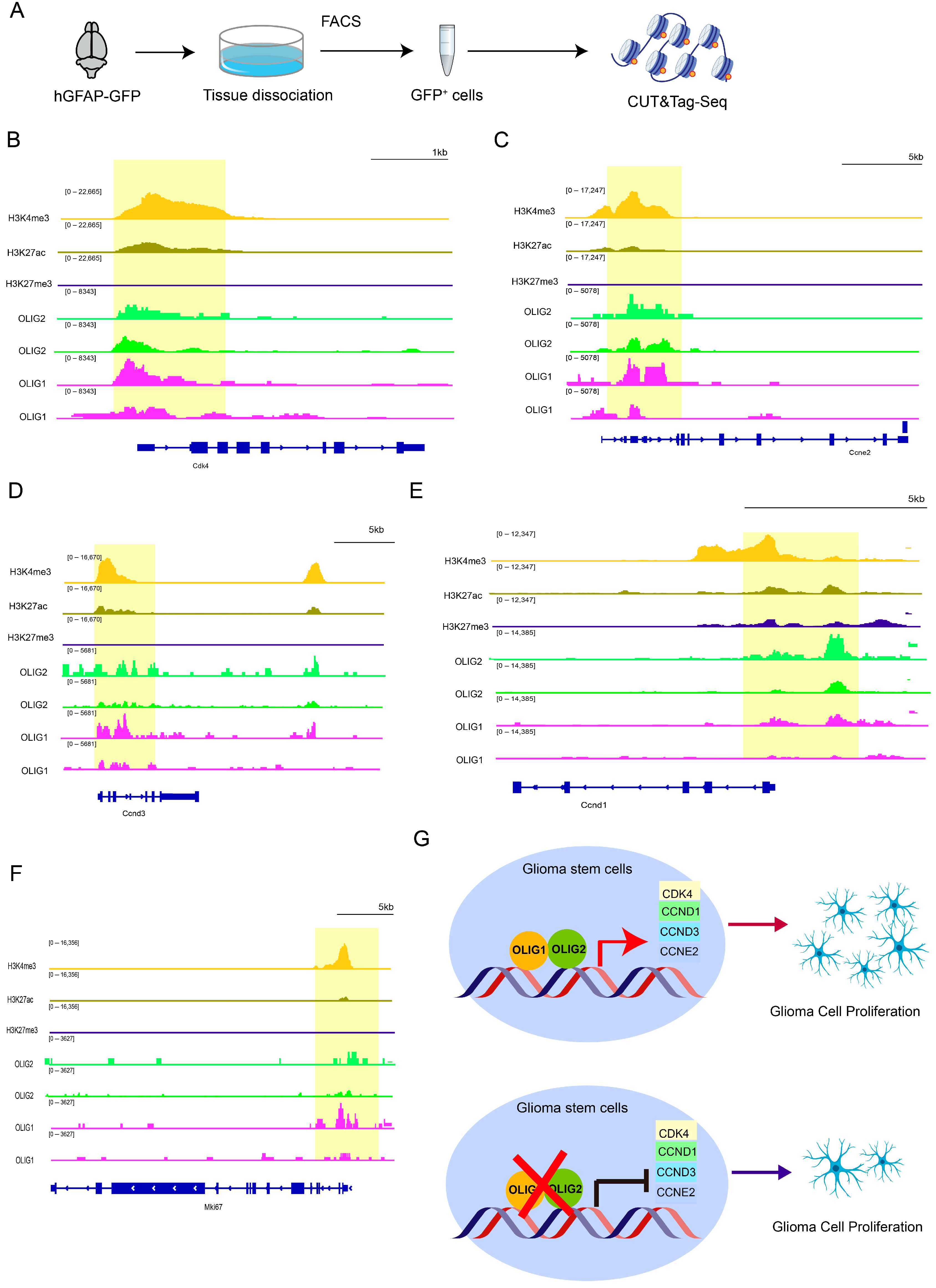
3.5. OLIG1/2 Promote Cell Proliferation by Binding to Various Cyclins
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Paes de Faria, J.; Kessaris, N.; Andrew, P.; Richardson, W.D.; Li, H. New Olig1 null mice confirm a non-essential role for Olig1 in oligodendrocyte development. BMC Neurosci. 2014, 15, 12. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, L.; Jiang, M.; Zhao, C.; Liu, X.; Berry, K.; Waisman, A.; Langseth, A.J.; Novitch, B.G.; Bergles, D.E.; et al. Olig2 Ablation in Immature Oligodendrocytes Does Not Enhance CNS Myelination and Remyelination. J. Neurosci. 2022, 42, 8542–8555. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, Y.; Kim, B.; Wang, H.; Zhao, C.; He, X.; Liu, L.; Liu, W.; Wu, L.M.; Mao, M.; et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 2013, 152, 248–261. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, S.; Yang, Q.; Guo, S.; Chen, Q.; Liu, Z.; Li, L.; Jiang, M.; Li, H.; Hu, J.; et al. The Oligodendrocyte Transcription Factor 2 OLIG2 regulates transcriptional repression during myelinogenesis in rodents. Nat. Commun. 2022, 13, 1423. [Google Scholar] [CrossRef]
- Lu, Q.R.; Sun, T.; Zhu, Z.; Ma, N.; Garcia, M.; Stiles, C.D.; Rowitch, D.H. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 2002, 109, 75–86. [Google Scholar] [CrossRef]
- Meijer, D.H.; Kane, M.F.; Mehta, S.; Liu, H.; Harrington, E.; Taylor, C.M.; Stiles, C.D.; Rowitch, D.H. Separated at birth? The functional and molecular divergence of OLIG1 and OLIG2. Nat. Rev. Neurosci. 2012, 13, 819–831. [Google Scholar] [CrossRef]
- Wegener, A.; Deboux, C.; Bachelin, C.; Frah, M.; Kerninon, C.; Seilhean, D.; Weider, M.; Wegner, M.; Nait-Oumesmar, B. Gain of Olig2 function in oligodendrocyte progenitors promotes remyelination. Brain 2015, 138, 120–135. [Google Scholar] [CrossRef]
- Zhou, Q.; Anderson, D.J. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell 2002, 109, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Breunig, J.J.; Levy, R.; Antonuk, C.D.; Molina, J.; Dutra-Clarke, M.; Park, H.; Akhtar, A.A.; Kim, G.B.; Hu, X.; Bannykh, S.I.; et al. Ets Factors Regulate Neural Stem Cell Depletion and Gliogenesis in Ras Pathway Glioma. Cell Rep. 2015, 12, 258–271. [Google Scholar] [CrossRef]
- Kupp, R.; Shtayer, L.; Tien, A.C.; Szeto, E.; Sanai, N.; Rowitch, D.H.; Mehta, S. Lineage-Restricted OLIG2-RTK Signaling Governs the Molecular Subtype of Glioma Stem-like Cells. Cell Rep. 2016, 16, 2838–2845. [Google Scholar] [CrossRef]
- Lu, Q.R.; Park, J.K.; Noll, E.; Chan, J.A.; Alberta, J.; Yuk, D.; Alzamora, M.G.; Louis, D.N.; Stiles, C.D.; Rowitch, D.H.; et al. Oligodendrocyte lineage genes (OLIG) as molecular markers for human glial brain tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 10851–10856. [Google Scholar] [CrossRef] [PubMed]
- Ligon, K.L.; Alberta, J.A.; Kho, A.T.; Weiss, J.; Kwaan, M.R.; Nutt, C.L.; Louis, D.N.; Stiles, C.D.; Rowitch, D.H. The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J. Neuropathol. Exp. Neurol. 2004, 63, 499–509. [Google Scholar] [CrossRef]
- Suvà, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Kosty, J.; Lu, F.; Kupp, R.; Mehta, S.; Lu, Q.R. Harnessing OLIG2 function in tumorigenicity and plasticity to target malignant gliomas. Cell Cycle 2017, 16, 1654–1660. [Google Scholar] [CrossRef]
- Liao, Y.; Luo, Z.; Deng, Y.; Zhang, F.; Rao, R.; Wang, J.; Xu, L.; Kumar, S.S.; Sengupta, S.; DeWire-Schottmiller, M.; et al. OLIG2 maintenance is not essential for diffuse intrinsic pontine glioma cell line growth but regulates tumor phenotypes. Neuro-Oncology 2021, 23, 1183–1196. [Google Scholar] [CrossRef]
- Ligon, K.L.; Huillard, E.; Mehta, S.; Kesari, S.; Liu, H.; Alberta, J.A.; Bachoo, R.M.; Kane, M.; Louis, D.N.; Depinho, R.A.; et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007, 53, 503–517. [Google Scholar] [CrossRef]
- Lu, F.; Chen, Y.; Zhao, C.; Wang, H.; He, D.; Xu, L.; Wang, J.; He, X.; Deng, Y.; Lu, E.E.; et al. Olig2-Dependent Reciprocal Shift in PDGF and EGF Receptor Signaling Regulates Tumor Phenotype and Mitotic Growth in Malignant Glioma. Cancer Cell 2016, 29, 669–683. [Google Scholar] [CrossRef]
- Wang, Z.; Tian, Y.; Fu, T.; Yang, F.; Li, J.; Yang, L.; Zhang, W.; Zheng, W.; Jiang, X.; Xu, Z.; et al. Coordinated regulation of cortical astrocyte maturation by OLIG1 and OLIG2 through BMP7 signaling modulation. J. Genet. Genom. 2025, in press. [CrossRef]
- Tian, Y.; Wang, Z.; Yang, F.; Zhang, W.; Li, J.; Yang, L.; Fu, T.; Zheng, W.; Xu, Z.; Ma, T.; et al. Orchestrating the Acquisition of Oligodendrocyte Precursor Cell versus Olfactory Bulb Interneuron Fates through Olig1/2 during Mammalian Cortical Gliogenesis and Gliomagenesis. bioRxiv 2024. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Verma, R.; Chen, X.; Xin, D.; Luo, Z.; Ogurek, S.; Xin, M.; Rao, R.; Berry, K.; Lu, Q.R. Olig1/2-Expressing Intermediate Lineage Progenitors Are Predisposed to PTEN/p53-Loss-Induced Gliomagenesis and Harbor Specific Therapeutic Vulnerabilities. Cancer Res. 2023, 83, 890–905. [Google Scholar] [CrossRef]
- Ligon, K.L.; Fancy, S.P.; Franklin, R.J.; Rowitch, D.H. Olig gene function in CNS development and disease. Glia 2006, 54, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Szu, J.I.; Tsigelny, I.F.; Wojcinski, A.; Kesari, S. Biological functions of the Olig gene family in brain cancer and therapeutic targeting. Front. Neurosci. 2023, 17, 1129434. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment. Annu. Rev. Pathol. 2014, 9, 1–25. [Google Scholar] [CrossRef]
- Li, J.L.; Yang, F.H.; Tian, Y.; Wang, Z.W.; Qi, D.S.; Yang, Z.G.; Song, J.G.; Ding, J.; Wang, X.; Zhang, Z.Z. Lateral/caudal ganglionic eminence makes limited contribution to cortical oligodendrocytes. eLife 2024, 13, RP94317. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Han, D.; Song, X.; Gao, Y.; Li, Z.; Li, X.; Yang, Z.; Xu, Z. Context-dependent regulation of Notch signaling in glial development and tumorigenesis. Sci. Adv. 2023, 9, eadi2167. [Google Scholar] [CrossRef]
- Li, Z.; Shang, Z.; Sun, M.; Jiang, X.; Tian, Y.; Yang, L.; Wang, Z.; Su, Z.; Liu, G.; Li, X.; et al. Transcription factor Sp9 is a negative regulator of D1-type MSN development. Cell Death Discov. 2022, 8, 301. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Wang, Z.; Lindtner, S.; Yang, L.; Shang, Z.; Tian, Y.; Guo, R.; You, Y.; Zhou, W.; Rubenstein, J.L.; et al. Dlx1/2-dependent expression of Meis2 promotes neuronal fate determination in the mammalian striatum. Development 2022, 149, dev200035. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Cai, Y.; Zhao, Z.; Shi, M.; Zheng, M.; Gong, L.; He, M. Embryonic origins of forebrain oligodendrocytes revisited by combinatorial genetic fate mapping. eLife 2024, 13, RP95406. [Google Scholar] [CrossRef]
- Yakovlev, A.G.; Faden, A.I. Caspase-dependent apoptotic pathways in CNS injury. Mol. Neurobiol. 2001, 24, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, G.; Yang, L.; Sun, M.; Zhang, Z.; Xu, Z.; Gao, Y.; Jiang, X.; Su, Z.; Li, X.; et al. BMP7 expression in mammalian cortical radial glial cells increases the length of the neurogenic period. Protein Cell 2024, 15, 21–35. [Google Scholar] [CrossRef]
- Shang, Z.; Yang, L.; Wang, Z.; Tian, Y.; Gao, Y.; Su, Z.; Guo, R.; Li, W.; Liu, G.; Li, X.; et al. The transcription factor Zfp503 promotes the D1 MSN identity and represses the D2 MSN identity. Front. Cell Dev. Biol. 2022, 10, 948331. [Google Scholar] [CrossRef]
- Wen, Y.; Su, Z.; Wang, Z.; Yang, L.; Liu, G.; Shang, Z.; Duan, Y.; Du, H.; Li, Z.; You, Y.; et al. Transcription Factor VAX1 Regulates the Regional Specification of the Subpallium Through Repressing Gsx2. Mol. Neurobiol. 2021, 58, 3729–3744. [Google Scholar] [CrossRef]
- Lee, S.K.; Lee, B.; Ruiz, E.C.; Pfaff, S.L. Olig2 and Ngn2 function in opposition to modulate gene expression in motor neuron progenitor cells. Genes Dev. 2005, 19, 282–294. [Google Scholar] [CrossRef]
- Li, H.; de Faria, J.P.; Andrew, P.; Nitarska, J.; Richardson, W.D. Phosphorylation regulates OLIG2 cofactor choice and the motor neuron-oligodendrocyte fate switch. Neuron 2011, 69, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Furusho, M.; Ono, K.; Takebayashi, H.; Masahira, N.; Kagawa, T.; Ikeda, K.; Ikenaka, K. Involvement of the Olig2 transcription factor in cholinergic neuron development of the basal forebrain. Dev. Biol. 2006, 293, 348–357. [Google Scholar] [CrossRef]
- Lu, Q.R.; Yuk, D.; Alberta, J.A.; Zhu, Z.; Pawlitzky, I.; Chan, J.; McMahon, A.P.; Stiles, C.D.; Rowitch, D.H. Sonic hedgehog–regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron 2000, 25, 317–329. [Google Scholar] [CrossRef]
- Tatsumi, K.; Isonishi, A.; Yamasaki, M.; Kawabe, Y.; Morita-Takemura, S.; Nakahara, K.; Terada, Y.; Shinjo, T.; Okuda, H.; Tanaka, T.; et al. Olig2-Lineage Astrocytes: A Distinct Subtype of Astrocytes That Differs from GFAP Astrocytes. Front. Neuroanat. 2018, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, K.; Kinugawa, K.; Isonishi, A.; Kitabatake, M.; Okuda, H.; Takemura, S.; Tanaka, T.; Mori, E.; Wanaka, A. Olig2-astrocytes express neutral amino acid transporter SLC7A10 (Asc-1) in the adult brain. Mol. Brain 2021, 14, 163. [Google Scholar] [CrossRef]
- Wang, H.; Xu, L.; Lai, C.; Hou, K.; Chen, J.; Guo, Y.; Sambangi, A.; Swaminathan, S.; Xie, C.; Wu, Z.; et al. Region-specific distribution of Olig2-expressing astrocytes in adult mouse brain and spinal cord. Mol. Brain 2021, 14, 36. [Google Scholar] [CrossRef]
- Marshall, C.A.; Novitch, B.G.; Goldman, J.E. Olig2 directs astrocyte and oligodendrocyte formation in postnatal subventricular zone cells. J. Neurosci. 2005, 25, 7289–7298. [Google Scholar] [CrossRef] [PubMed]
- Myers, B.L.; Brayer, K.J.; Paez-Beltran, L.E.; Villicana, E.; Keith, M.S.; Suzuki, H.; Newville, J.; Anderson, R.H.; Lo, Y.; Mertz, C.M.; et al. Transcription factors ASCL1 and OLIG2 drive glioblastoma initiation and co-regulate tumor cell types and migration. Nat. Commun. 2024, 15, 10363. [Google Scholar] [CrossRef]
- Zhang, L.; He, X.; Liu, X.; Zhang, F.; Huang, L.F.; Potter, A.S.; Xu, L.; Zhou, W.; Zheng, T.; Luo, Z.; et al. Single-Cell Transcriptomics in Medulloblastoma Reveals Tumor-Initiating Progenitors and Oncogenic Cascades during Tumorigenesis and Relapse. Cancer Cell 2019, 36, 302–318.e7. [Google Scholar] [CrossRef]
- Zou, F.; Xie, G.; Ma, J.A.; Zhou, D.A.; Jiang, Y.I.; Zheng, J.Y. Epidermal growth factor receptor mutation heterogeneity analysis of pulmonary sarcomatoid carcinoma successfully treated with erlotinib: A case report. Oncol. Lett. 2015, 9, 2239–2243. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Wang, Z.; Sun, M.; Li, J.; Zheng, W.; Yang, F.; Zhang, Z. Olig1/2 Drive Astrocytic Glioblastoma Proliferation Through Transcriptional Co-Regulation of Various Cyclins. Genes 2025, 16, 573. https://doi.org/10.3390/genes16050573
Tian Y, Wang Z, Sun M, Li J, Zheng W, Yang F, Zhang Z. Olig1/2 Drive Astrocytic Glioblastoma Proliferation Through Transcriptional Co-Regulation of Various Cyclins. Genes. 2025; 16(5):573. https://doi.org/10.3390/genes16050573
Chicago/Turabian StyleTian, Yu, Ziwu Wang, Mengge Sun, Jialin Li, Wenhui Zheng, Feihong Yang, and Zhuangzhi Zhang. 2025. "Olig1/2 Drive Astrocytic Glioblastoma Proliferation Through Transcriptional Co-Regulation of Various Cyclins" Genes 16, no. 5: 573. https://doi.org/10.3390/genes16050573
APA StyleTian, Y., Wang, Z., Sun, M., Li, J., Zheng, W., Yang, F., & Zhang, Z. (2025). Olig1/2 Drive Astrocytic Glioblastoma Proliferation Through Transcriptional Co-Regulation of Various Cyclins. Genes, 16(5), 573. https://doi.org/10.3390/genes16050573