
Genetic Background and Gene Essentiality


Abstract
1. Introduction
2. Materials and Methods
2.1. C. elegans Strains
2.2. Query Genes
2.3. RNA Interference and Bacterial Preparation
2.4. Worm Preparation
2.5. Fitness Assay
2.6. Assay Quality Control
- (i)
- Initial OD between 0.745 and 0.905;
- (ii)
- Worm density between 10 and 30 individuals per well;
- (iii)
- Absence of contamination with bacteria or fungi, as assessed visually.
2.7. Data Standardization
2.8. Data Quality Control
2.9. Statistical Analysis
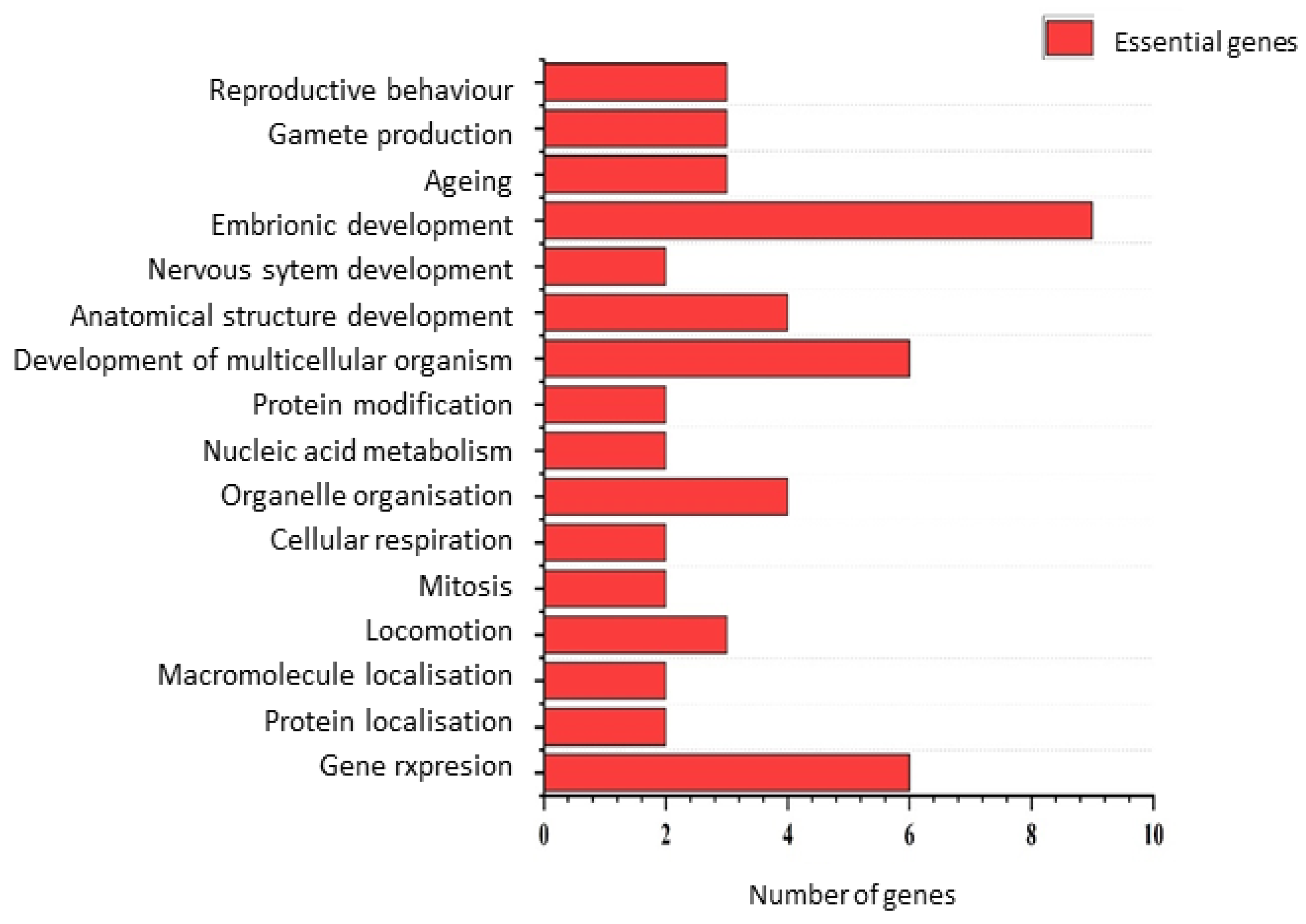
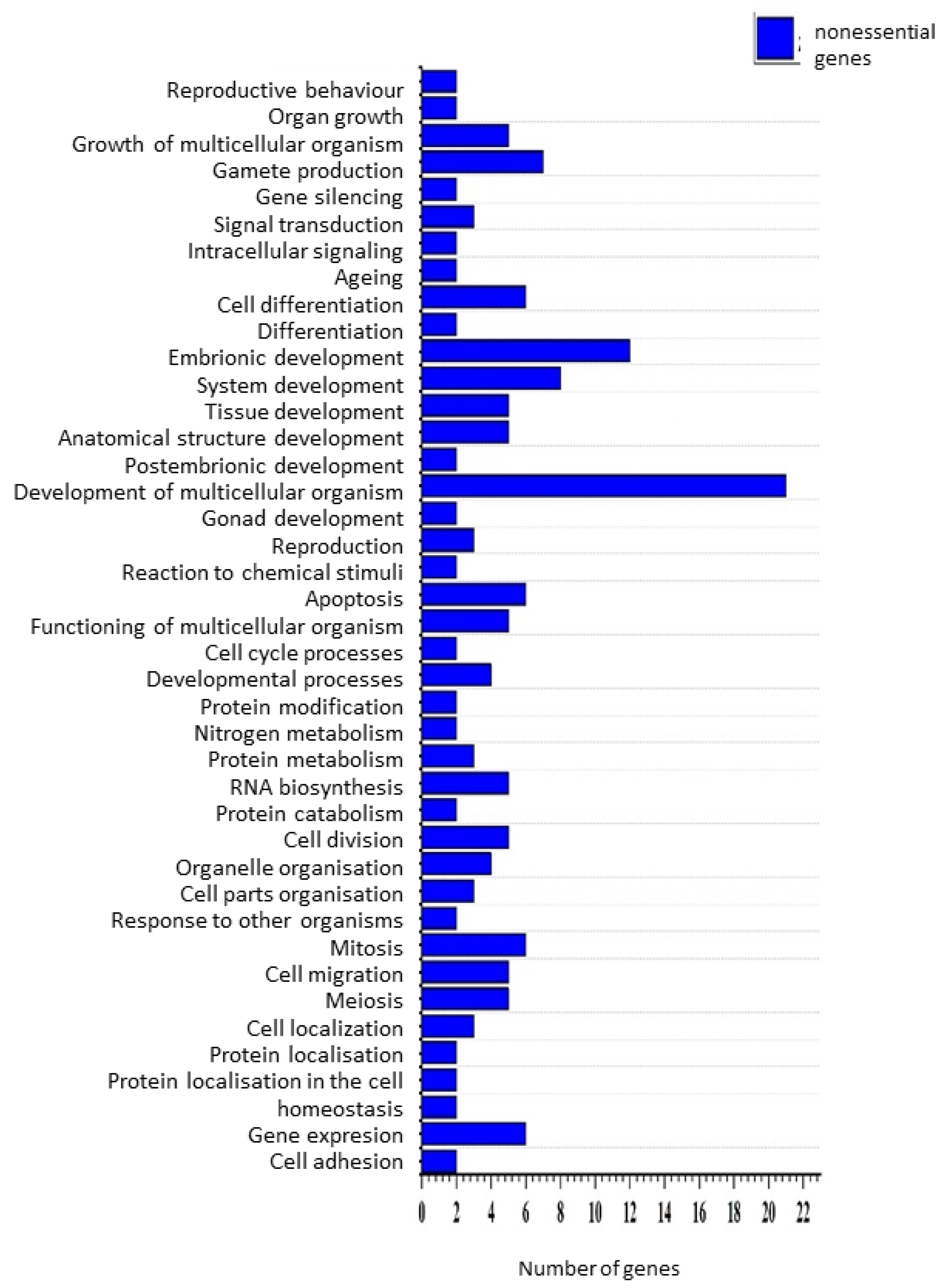
2.10. Gene Ontology
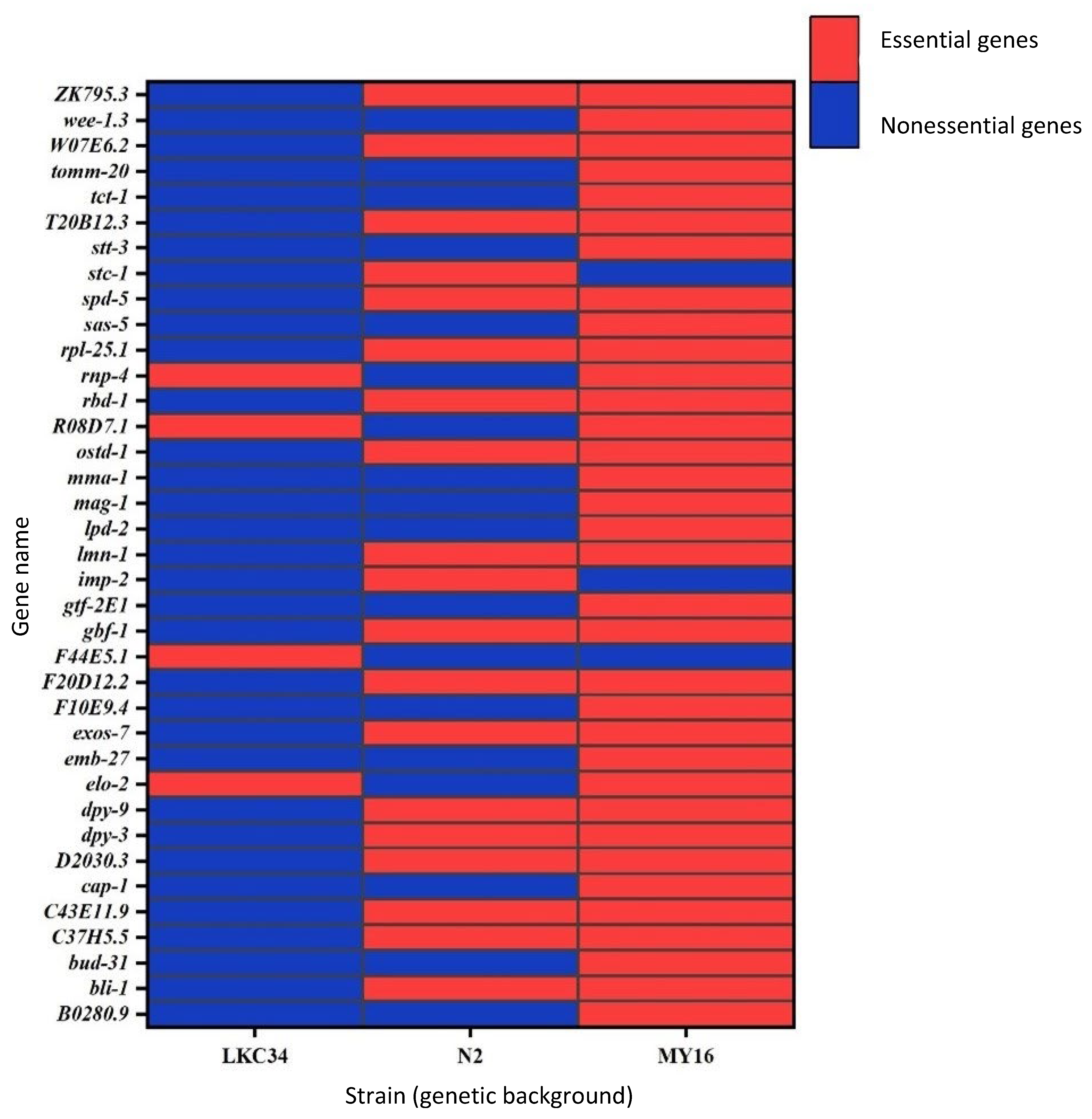
3. Results
- Statistical significance: Genes were classified as essential if their fitness was significantly lower than that of wild-type worms (adjusted p < 0.01 from a t-test or Wilcoxon test). This method showed 58% agreement with OGEE (Supplementary Table S1).
- Fitness threshold I: Genes with an average fitness <0.9 of wild-type fitness were considered essential. This method yielded 68% agreement with OGEE (Supplementary Table S1).
- Fitness threshold II: Genes with an average fitness <0.75 of wild-type fitness were considered essential. This criterion produced the highest agreement with OGEE at 70%. We adopted this method for all subsequent analyses.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gluecksohn-Waelsch, S. Lethal Genes and Analysis of Differentiation: In higher organisms lethal genes serve as tools for studies of cell differentiation and cell genetics. Science 1963, 142, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Fraser, C.M.; Gocayne, J.D.; White, O.; Adams, M.D.; Clayton, R.A.; Fleischmann, R.D.; Bult, C.J.; Kerlavage, A.R.; Sutton, G.; Kelley, J.M. The minimal gene complement of Mycoplasma genitalium. Science 1995, 270, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.-F.; Dougherty, B.A.; Merrick, J.M. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, C.A., III; Peterson, S.N.; Gill, S.R.; Cline, R.T.; White, O.; Fraser, C.M.; Smith, H.O.; Craig Venter, J. Global transposon mutagenesis and a minimal Mycoplasma genome. Science 1999, 286, 2165–2169. [Google Scholar] [CrossRef]
- Luo, H.; Lin, Y.; Liu, T.; Lai, F.-L.; Zhang, C.-T.; Gao, F.; Zhang, R. DEG 15, an update of the Database of Essential Genes that includes built-in analysis tools. Nucl. Acid. Res. 2021, 49, D677–D686. [Google Scholar] [CrossRef]
- Rancati, G.; Moffat, J.; Typas, A.; Pavelka, N. Emerging and evolving concepts in gene essentiality. Nat. Rev. Genet. 2018, 19, 34–49. [Google Scholar] [CrossRef]
- Gerdes, S.; Scholle, M.; Campbell, J.; Balazsi, G.; Ravasz, E.; Daugherty, M.; Somera, A.; Kyrpides, N.; Anderson, I.; Gelfand, M. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J. Bacteriol. 2003, 185, 5673–5684. [Google Scholar] [CrossRef]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2, 2006.0008. [Google Scholar] [CrossRef]
- Paaby, A.B.; White, A.G.; Riccardi, D.D.; Gunsalus, K.C.; Piano, F.; Rockman, M.V. Wild worm embryogenesis harbors ubiquitous polygenic modifier variation. elife 2015, 4, e09178. [Google Scholar] [CrossRef]
- Nichols, R.J.; Sen, S.; Choo, Y.J.; Beltrao, P.; Zietek, M.; Chaba, R.; Lee, S.; Kazmierczak, K.M.; Lee, K.J.; Wong, A. Phenotypic landscape of a bacterial cell. Cell 2011, 144, 143–156. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Ussery, D.W.; Wassenaar, T.M. Genome update: The 1000th genome—A cautionary tale. Microbiology 2010, 156, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Stiernagle, T. Maintenance of C. elegans. In WormBook: The Online Review of C. elegans Biology; WormBook: Pasadena, CA, USA, 2006. [Google Scholar]
- Kamath, R.S.; Fraser, A.G.; Dong, Y.; Poulin, G.; Durbin, R.; Gotta, M.; Kanapin, A.; Le Bot, N.; Moreno, S.; Sohrmann, M. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 2003, 421, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-H.; Minguez, P.; Lercher, M.J.; Bork, P. OGEE: An online gene essentiality database. Nucl. Acid. Res. 2012, 40, D901–D906. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Elvin, M.; Snoek, L.B.; Frejno, M.; Klemstein, U.; Kammenga, J.E.; Poulin, G.B. A fitness assay for comparing RNAi effects across multiple C. elegans genotypes. BMC Genom. 2011, 12, 510. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Consortium, T.G.O. The Gene Ontology resource: Enriching a GOld mine. Nucl. Acid. Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Davis, P.; Zarowiecki, M.; Arnaboldi, V.; Becerra, A.; Cain, S.; Chan, J.; Chen, W.J.; Cho, J.; da Veiga Beltrame, E.; Diamantakis, S. WormBase in 2022—Data, processes, and tools for analyzing Caenorhabditis elegans. Genetics 2022, 220, iyac003. [Google Scholar] [CrossRef]
- Ramani, A.K.; Chuluunbaatar, T.; Verster, A.J.; Na, H.; Vu, V.; Pelte, N.; Wannissorn, N.; Jiao, A.; Fraser, A.G. The majority of animal genes are required for wild-type fitness. Cell 2012, 148, 792–802. [Google Scholar] [CrossRef]
- Vu, V.; Verster, A.J.; Schertzberg, M.; Chuluunbaatar, T.; Spensley, M.; Pajkic, D.; Hart, G.T.; Moffat, J.; Fraser, A.G. Natural variation in gene expression modulates the severity of mutant phenotypes. Cell 2015, 162, 391–402. [Google Scholar] [CrossRef]
- Andersen, E.C.; Gerke, J.P.; Shapiro, J.A.; Crissman, J.R.; Ghosh, R.; Bloom, J.S.; Félix, M.-A.; Kruglyak, L. Chromosome-scale selective sweeps shape Caenorhabditis elegans genomic diversity. Nat. Genet. 2012, 44, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F. Epistasis and quantitative traits: Using model organisms to study gene–gene interactions. Nat. Rev. Genet. 2014, 15, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.B.; Ehrenreich, I.M. Higher-order genetic interactions and their contribution to complex traits. Trends Genet. 2015, 31, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Sterken, M.G.; Snoek, L.B.; Kammenga, J.E.; Andersen, E.C. The laboratory domestication of Caenorhabditis elegans. Trends Genet. 2015, 31, 224–231. [Google Scholar] [CrossRef]
- Blomen, V.A.; Májek, P.; Jae, L.T.; Bigenzahn, J.W.; Nieuwenhuis, J.; Staring, J.; Sacco, R.; van Diemen, F.R.; Olk, N.; Stukalov, A. Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350, 1092–1096. [Google Scholar] [CrossRef]
- Yu, S.; Zheng, C.; Zhou, F.; Baillie, D.L.; Rose, A.M.; Deng, Z.; Chu, J.S.-C. Genomic identification and functional analysis of essential genes in Caenorhabditis elegans. BMC Genom. 2018, 19, 871. [Google Scholar] [CrossRef]
- Bartha, I.; Di Iulio, J.; Venter, J.C.; Telenti, A. Human gene essentiality. Nat. Rev. Genet. 2018, 19, 51–62. [Google Scholar] [CrossRef]
- Zhang, Z.; Ren, Q. Why are essential genes essential?—The essentiality of Saccharomyces genes. Microb. Cell 2015, 2, 280. [Google Scholar] [CrossRef]
- Hutchison, C.A., III; Chuang, R.-Y.; Noskov, V.N.; Assad-Garcia, N.; Deerinck, T.J.; Ellisman, M.H.; Gill, J.; Kannan, K.; Karas, B.J.; Ma, L. Design and synthesis of a minimal bacterial genome. Science 2016, 351, aad6253. [Google Scholar] [CrossRef]
- Roemer, T.; Jiang, B.; Davison, J.; Ketela, T.; Veillette, K.; Breton, A.; Tandia, F.; Linteau, A.; Sillaots, S.; Marta, C.; et al. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol. Microbiol. 2003, 50, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Ge, X.; Chen, L.; Wang, X.; Dou, Y.; Xu, J.Z.; Patel, J.R.; Stone, V.; Trinh, M.; Evans, K.; et al. Genome-wide essential gene identification in Streptococcus sanguinis. Sci. Rep. 2011, 1, 125. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhan, T.; Boutros, M. Towards a compendium of essential genes—From model organisms to synthetic lethality in cancer cells. Crit. Rev. Biochem. Mol. Biol. 2015, 51, 74–85. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | df | F-Value | p-Value |
|---|---|---|---|
| Strain | 2 | 3547.2 | <0.001 |
| Mutation | 293 | 469.8 | <0.001 |
| Strain × Mutation | 586 | 34.3 | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gąsienica, P.; Toch, K.; Zając-Garlacz, K.S.; Labocha-Derkowska, M. Genetic Background and Gene Essentiality. Genes 2025, 16, 570. https://doi.org/10.3390/genes16050570
Gąsienica P, Toch K, Zając-Garlacz KS, Labocha-Derkowska M. Genetic Background and Gene Essentiality. Genes. 2025; 16(5):570. https://doi.org/10.3390/genes16050570
Chicago/Turabian StyleGąsienica, Paulina, Katarzyna Toch, Kamila Stefania Zając-Garlacz, and Marta Labocha-Derkowska. 2025. "Genetic Background and Gene Essentiality" Genes 16, no. 5: 570. https://doi.org/10.3390/genes16050570
APA StyleGąsienica, P., Toch, K., Zając-Garlacz, K. S., & Labocha-Derkowska, M. (2025). Genetic Background and Gene Essentiality. Genes, 16(5), 570. https://doi.org/10.3390/genes16050570