

Mitochondrial Genomes of the Robberflies Clephydroneura jiangxiensis and Maira xizangensis (Diptera: Asilidae) and Phylogeny of Three Superfamilies

Abstract
1. Introduction
2. Methods
2.1. Taxa Sampled and DNA Extraction
2.2. Data Collection
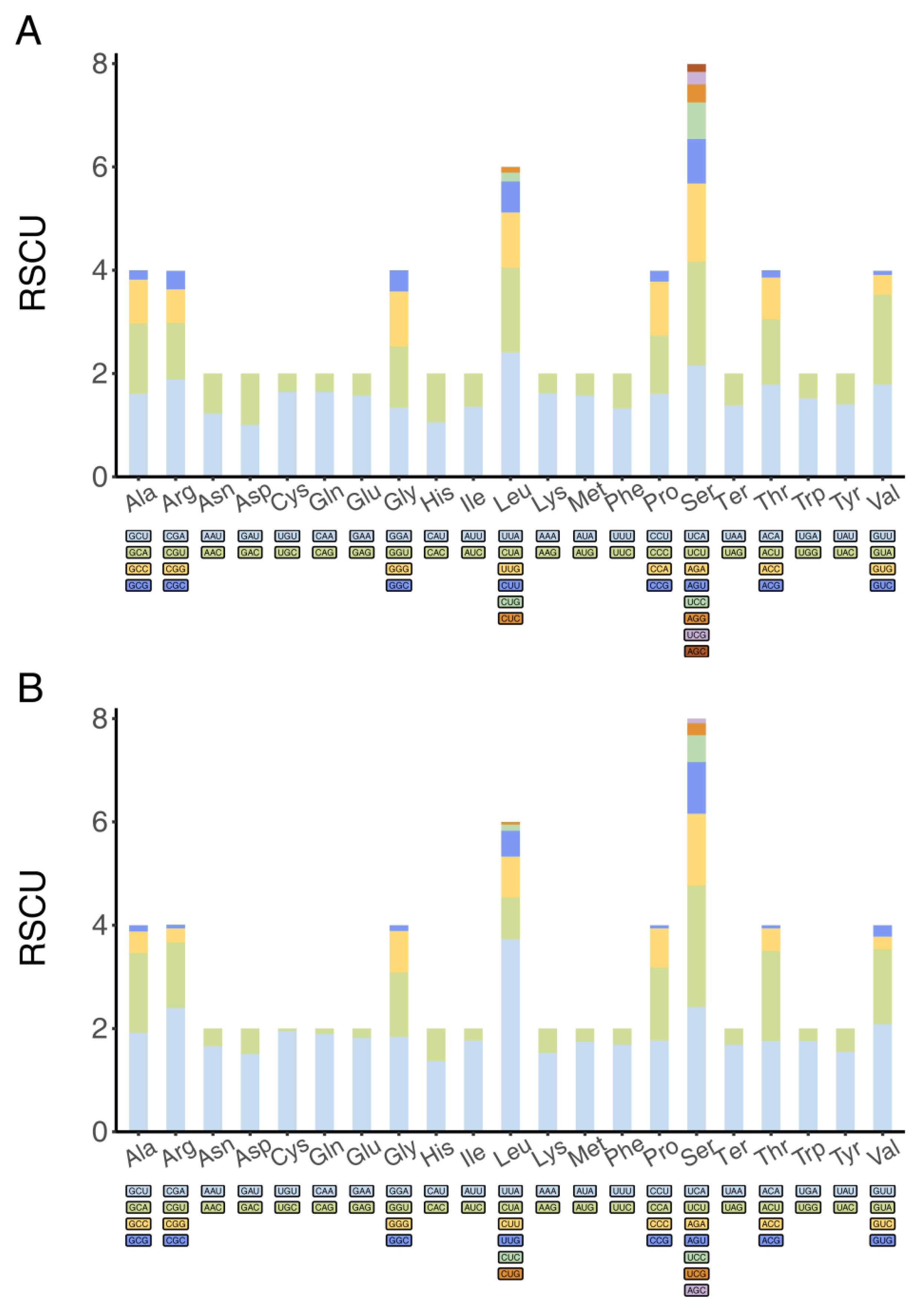
2.3. Gene and Codon Analysis
2.4. Phylogeny Estimation
3. Results
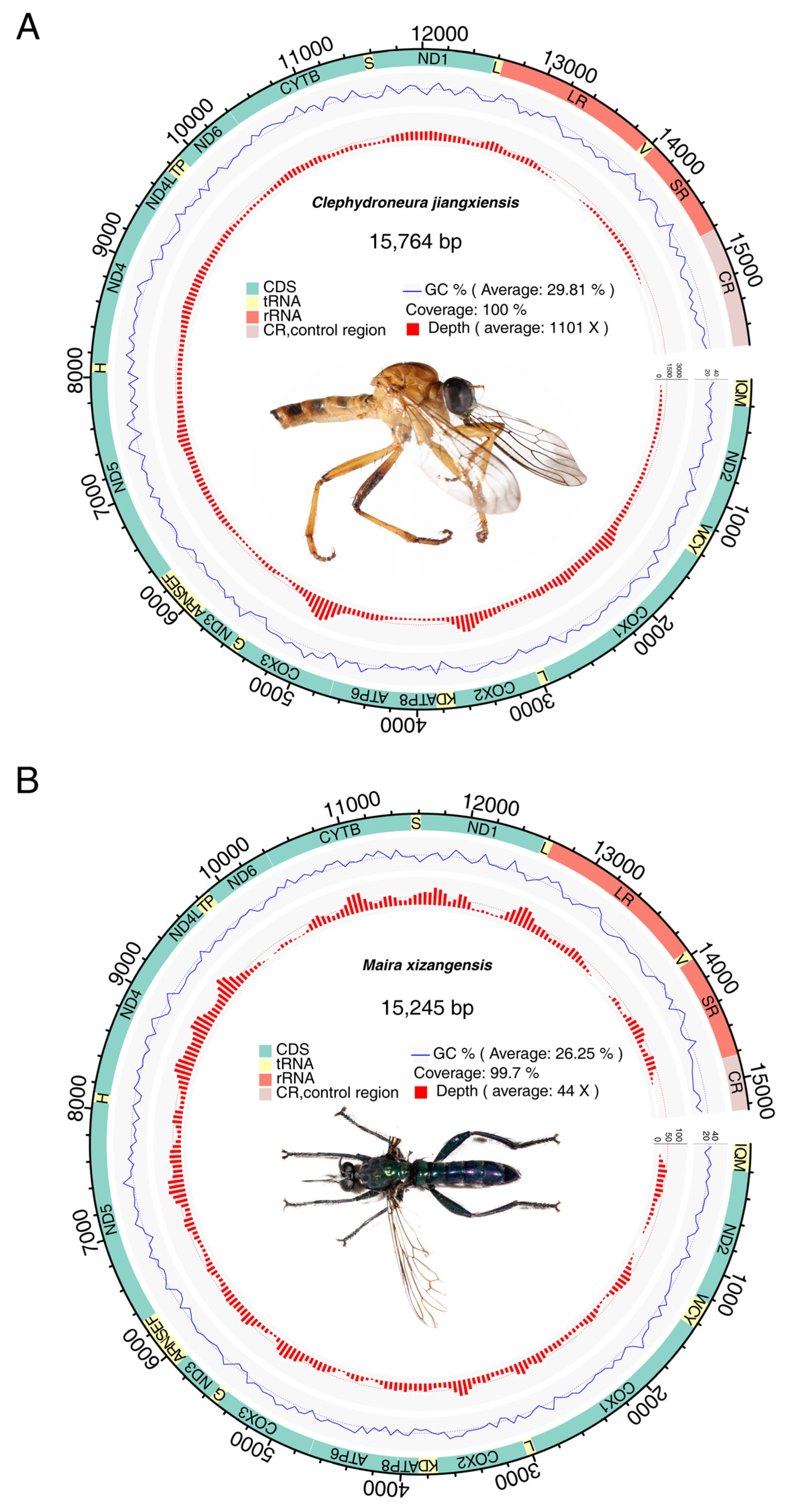
3.1. Basic Features of Mitochondrial Genomes
3.2. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rohdendorf, B.B. The Historical Development of Diptera; University of Alberta Press: Edmonton, AB, Canada, 1974. [Google Scholar]
- Grimaldi, D.A.; Arillo, A.; Cumming, J.M.; Hauser, M. Brachyceran Diptera (Insecta) in Cretaceous Ambers, Part IV: Significant New Orthorrhaphous Taxa. ZooKeys 2011, 148, 293–332. [Google Scholar] [CrossRef] [PubMed]
- Ren, D. Flower-Associated Brachycera Flies as Fossil Evidence for Jurassic Angiosperm Origins. Science 1998, 280, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Marchiori, C.H. Study of the Biological and Taxonomic Characteristics of the Families Megamerinidae, Nemestrinidae and Therevidae (Insecta: Diptera). Open Access Res. J. Biol. Pharm. 2023, 7, 043–074. [Google Scholar] [CrossRef]
- Marston, N. The Biology of Anthrax Limatulus Fur (Osten Sacken), with a Key to and Descriptions of Pupae of Some Species in the Anthrax Albofasciatus and Trimaculatus Groups (Diptera: Bombyliidae). J. Kans. Entomol. Soc. 1964, 37, 89–105. [Google Scholar]
- Bohart, G.E.; Stephen, W.P.; Eppley, R.K. The Biology of Heterostylum Robustum (Diptera: Bombyliidae), A Parasite of the Alkali Bee. Ann. Entomol. Soc. Am. 1960, 53, 425–435. [Google Scholar] [CrossRef]
- Greathead, D.J. Observations on Two Species of Systoechus (Diptera: Bombyliidae) Preying on the Desert Locust, Schistocerca Gregaria (Forskål), in Eastern Africa. Entomophaga 1958, 3, 3–22. [Google Scholar] [CrossRef]
- Rohdendorf, B.B.; Davis, D.R. Fundamentals of Paleontology; Smithsonian Institution Libraries and National Science Foundation: Washington, DC, USA; Alexandria, VA, USA, 1991. [Google Scholar]
- Bei-Bienko, G.Y. Congrès International D’entomologie: [Proceedings]. In Proceedings of the 1st International Congress of Entomology; 1910, 1, 294. Brussels, Belgium: International Entomological Society. Available online: https://www.biodiversitylibrary.org/page/58371224 (accessed on 5 May 2025).
- Dikow, T. Phylogeny of Asilidae Inferred from Morphological Characters of Imagines (Insecta: Diptera: Brachycera: Asiloidea). Bull. Am. Mus. Nat. Hist. 2009, 319, 1–175. [Google Scholar] [CrossRef]
- Trautwein, M.D.; Wiegmann, B.M.; Yeates, D.K. A Multigene Phylogeny of the Fly Superfamily Asiloidea (Insecta): Taxon Sampling and Additional Genes Reveal the Sister-Group to All Higher Flies (Cyclorrhapha). Mol. Phylogenet. Evol. 2010, 56, 918–930. [Google Scholar] [CrossRef]
- Amorim, D.d.S.; Silva, V.C.; Brown, B.V. Puyehuemyia Chandleri, Gen. Nov., Sp. Nov. (Diptera, Opetiidae): Remnant of a Cretaceous Biota in Chile. Am. Mus. Nat. Hist. 2018, 3892, 1–27. [Google Scholar]
- Wiegmann, B.M.; Trautwein, M.D.; Winkler, I.S.; Barr, N.B.; Kim, J.-W.; Lambkin, C.; Bertone, M.A.; Cassel, B.K.; Bayless, K.M.; Heimberg, A.M.; et al. Episodic Radiations in the Fly Tree of Life. Proc. Natl. Acad. Sci. USA 2011, 108, 5690–5695. [Google Scholar] [CrossRef]
- Kotrba, M. Bonner Zoologische Monographien; Zoologisches Forschungsinstitut und Museum Alexander Koenig: Munich, Germany, 1993. [Google Scholar]
- Yeates, D.K. The Cladistics and Classification of the Bombyliidae (Diptera: Asiloidea). Bull. Am. Mus. Nat. Hist. 1994, 219, 1–191. [Google Scholar]
- Shin, S.; Bayless, K.M.; Winterton, S.L.; Dikow, T.; Lessard, B.D.; Yeates, D.K.; Wiegmann, B.M.; Trautwein, M.D. Taxon Sampling to Address an Ancient Rapid Radiation: A Supermatrix Phylogeny of Early Brachyceran Flies (Diptera). Syst. Entomol. 2018, 43, 277–289. [Google Scholar] [CrossRef]
- Yao, G.; An, Y.; Luo, J.; Zhang, Z.; Yang, D.; Wang, Y. Mitochondrial Genomes of Bombyliidae (Diptera): Phylogenetic Analysis Recovers Monophyletic Bombyliidae Sister to Asilidae. Eur. J. Entomol. 2023, 120, 349–356. [Google Scholar] [CrossRef]
- Kahanpää, J. Checklist of the Empidoidea of Finland (Insecta, Diptera). ZooKeys 2014, 441, 183–207. [Google Scholar] [CrossRef]
- Sinclair, B.J.; Cumming, J.M. The Morphology, Higher-Level Phylogeny and Classification of the Empidoidea (Diptera). Zootaxa 2006, 1180, 1–172. [Google Scholar] [CrossRef]
- Grichanov, I.Y. An Illustrated Synopsis and Keys to Afrotropical Genera of the Epifamily Dolichopodoidae (Diptera: Empidoidea). Priamus suppl. 2011, 24, 1–98. [Google Scholar]
- Droop, A.P. Fqtools: An Efficient Software Suite for Modern FASTQ File Manipulation. Bioinformatics 2016, 32, 1883–1884. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A Toolkit for Animal Mitochondrial Genome Assembly, Annotation and Visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect Mitochondrial Genomics: Implications for Evolution and Phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize Implements and Enhances Circular Visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Xi, Y.Q.; Yin, X.M. Phylogenetic Relationships of Brachycera (Insecta: Diptera) Inferred from Mitochondrial Genome Sequences. Zool. J. Linn. Soc. 2022, 196, 720–739. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. Codon Usage in Regulatory Genes in Escherichia coli Does Not Reflect Selection for ‘Rare’ Codons. Nucleic Acids Res. 1986, 14, 7737–7749. [Google Scholar] [CrossRef]
- Gómez-Rubio, V. Ggplot2-Elegant Graphics for Data Analysis (2nd Edition). J. Stat. Soft. 2017, 77, 1–3. [Google Scholar] [CrossRef]
- Zhang, Z. KaKs_Calculator 3.0: Calculating Selective Pressure on Coding and Non-Coding Sequences. Genom. Proteom. Bioinform. 2022, 20, 536–540. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. PartitionFinder: Combined Selection of Partitioning Schemes and Substitution Models for Phylogenetic Analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, P.; Rieb, E.; Abrami, G.; Lücking, A.; Mehler, A. TREEANNOTATOR: Versatile Visual Annotation of Hierarchical Text Relations. In Proceedings of the Eleventh International Conference on Language Resources and Evaluation (LREC 2018), Miyazaki, Japan, 7–12 May 2018; pp. 1958–1963. [Google Scholar]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. GGTREE: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Wolstenholme, D.R.; Clary, D.O. Sequence Evolution of Drosophila Mitochondrial DNA. Genetics 1985, 109, 725–744. [Google Scholar] [CrossRef]
- Wang, G.; Huang, M. Characterization of the Complete Mitochondrial Genome of Simulium (Byssodon) maculatum (Diptera: Simuliidae) and Its Phylogenetic Implications. Int. J. Biol. Macromol. 2019, 121, 152–160. [Google Scholar] [CrossRef]
- Lei, T.; Zheng, X.; Song, C.; Jin, H.; Chen, L.; Qi, X. Limited Variation in Codon Usage across Mitochondrial Genomes of Non-Biting Midges (Diptera: Chironomidae). Insects 2024, 15, 752. [Google Scholar] [CrossRef]
- Percudani, R.; Pavesi, A.; Ottonello, S. Transfer RNA Gene Redundancy and Translational Selection in Saccharomyces cerevisiae. J. Mol. Biol. 1997, 268, 322–330. [Google Scholar] [CrossRef]
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An Index of Substitution Saturation and Its Application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Hennig, W. Flügelgeäder Und System Der Dipteren Unter Besonderer Berücksichtigung Der Aus Dem Mesozoikum Beschriebenen Fossilien. Beiträge Zur Entomol. 1954, 4, 245–388. [Google Scholar] [CrossRef]
- Hennig, W. Diptera (Zweiflügler). In Handbuch Der Zoologie, Band IV Arthropoda–Insecta; De Gruyter: Berlin, Germany, 1973. [Google Scholar]
- Woodley, N.E. Phylogeny and Classification of the “Orthorrhaphous” Brachycera. Man. Near. Dipt. 1989, 3, 1371–1395. [Google Scholar]
- Wiegmann, B.M.; Tsaur, S.C.; Webb, D.W.; Yeates, D.K.; Cassel, B.K. Monophyly and Relationships of the Tabanomorpha (Diptera: Brachycera) Based on 28S Ribosomal Gene Sequences. Ann. Èntomol. Soc. Am. 2000, 93, 1031–1038. [Google Scholar] [CrossRef]
- Wiegmann, B.M.; Yeates, D.K.; Thorne, J.L.; Kishino, H. Time Flies, a New Molecular Time-Scale for Brachyceran Fly Evolution without a Clock. Syst. Biol. 2003, 52, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Yeates, D.K. Relationships of Extant Lower Brachycera (Diptera): A Quantitative Synthesis of Morphological Characters. Zool. Scr. 2002, 31, 105–121. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | A (%) | C (%) | G (%) | T (%) | AT (%) | Size (bp) | Proportion in the Genome (%) |
|---|---|---|---|---|---|---|---|
| C. jiangxiensis | |||||||
| genome | 41.5 | 19.5 | 10.3 | 28.7 | 70.2 | 15,764 | 100 |
| PCGs | 40.3 | 21.5 | 11.3 | 26.9 | 67.2 | 11,212 | 71.1 |
| rRNA | 45.2 | 16.6 | 7.6 | 30.7 | 75.9 | 2084 | 13.2 |
| tRNA | 40.9 | 14.7 | 11.1 | 33.2 | 74.1 | 1473 | 9.3 |
| Control region | 47.1 | 10.2 | 4.3 | 38.4 | 85.5 | 943 | 6.0 |
| M. xizangensis | |||||||
| genome | 39.2 | 16.2 | 10.1 | 34.6 | 73.8 | 15,245 | 100 |
| PCGs | 38.0 | 17.2 | 10.7 | 34.1 | 72.1 | 11,230 | 73.7 |
| rRNA | 43.4 | 14.8 | 8.0 | 33.8 | 77.2 | 2104 | 13.8 |
| tRNA | 39.4 | 13.7 | 10.7 | 36.3 | 75.7 | 1463 | 9.6 |
| Control region | 47.9 | 5.7 | 1.7 | 44.8 | 92.7 | 422 | 2.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, K.; Lu, J.; Xu, S.-Q. Mitochondrial Genomes of the Robberflies Clephydroneura jiangxiensis and Maira xizangensis (Diptera: Asilidae) and Phylogeny of Three Superfamilies. Genes 2025, 16, 561. https://doi.org/10.3390/genes16050561
Zhang K, Lu J, Xu S-Q. Mitochondrial Genomes of the Robberflies Clephydroneura jiangxiensis and Maira xizangensis (Diptera: Asilidae) and Phylogeny of Three Superfamilies. Genes. 2025; 16(5):561. https://doi.org/10.3390/genes16050561
Chicago/Turabian StyleZhang, Keyao, Junhui Lu, and Sheng-Quan Xu. 2025. "Mitochondrial Genomes of the Robberflies Clephydroneura jiangxiensis and Maira xizangensis (Diptera: Asilidae) and Phylogeny of Three Superfamilies" Genes 16, no. 5: 561. https://doi.org/10.3390/genes16050561
APA StyleZhang, K., Lu, J., & Xu, S.-Q. (2025). Mitochondrial Genomes of the Robberflies Clephydroneura jiangxiensis and Maira xizangensis (Diptera: Asilidae) and Phylogeny of Three Superfamilies. Genes, 16(5), 561. https://doi.org/10.3390/genes16050561