
ACADVL Deep Sequencing in a Case Study: Beyond the Common c.848T>C Pathogenic Variant

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Enrollment
2.2. Newborn Screening and Confirmatory Testing
2.3. ACADVL Deep Sequencing
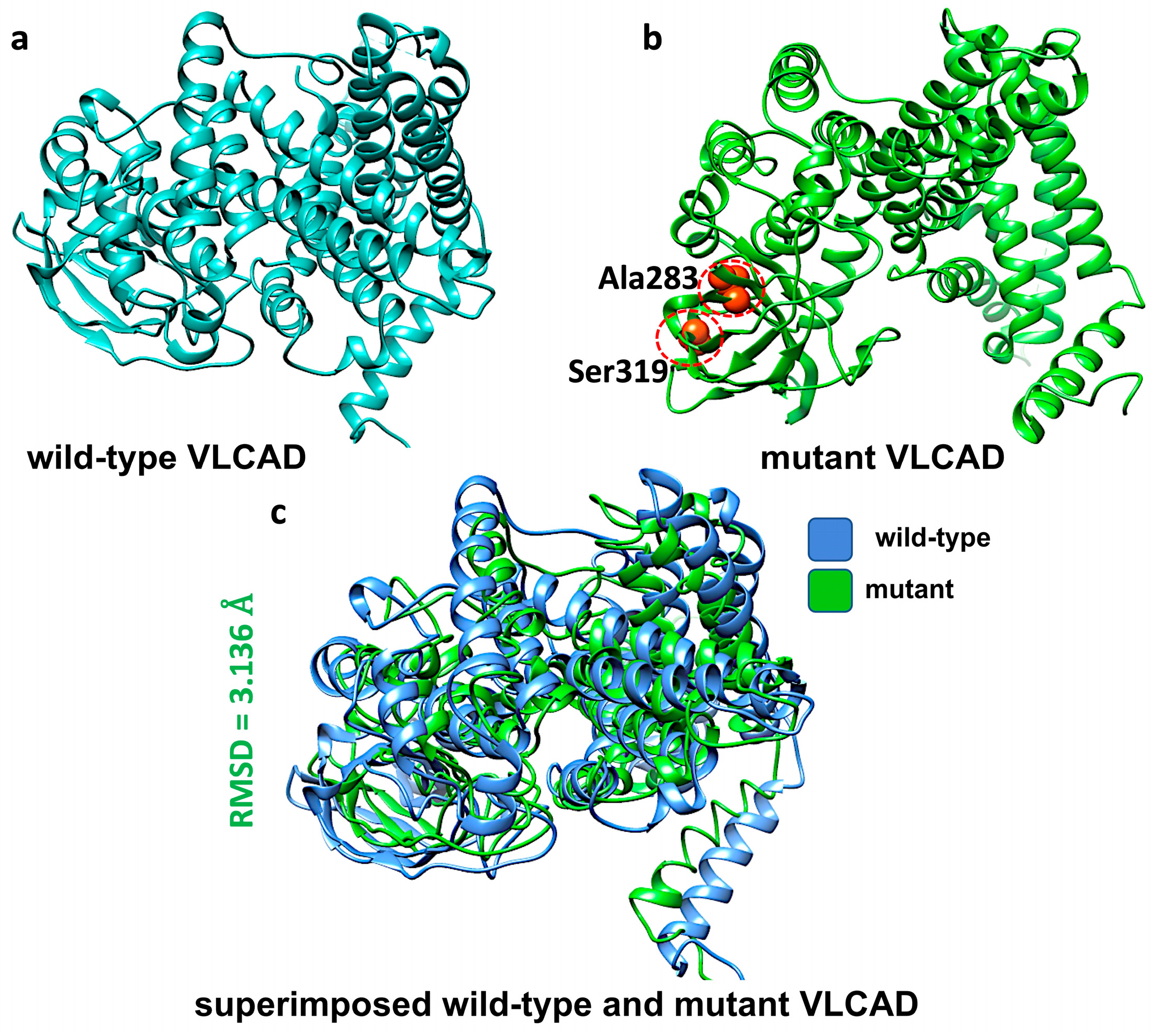
2.4. Protein Molecular Modelling
2.5. ACADVL RNA Evaluation
3. Results
3.1. Clinical Report
3.2. Genetic and Enzymatic Assessment
3.3. In Silico Characterization of the Variants
3.4. ACADVL Synonymous Variants
3.5. Gene Expression Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Navarrete, R.; Leal, F.; Vega, A.I.; Morais-López, A.; Garcia-Silva, M.T.; Martín-Hernández, E.; Quijada-Fraile, P.; Bergua, A.; Vives, I.; García-Jiménez, I.; et al. Value of Genetic Analysis for Confirming Inborn Errors of Metabolism Detected through the Spanish Neonatal Screening Program. Eur. J. Hum. Genet 2019, 27, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Ruoppolo, M.; Malvagia, S.; Boenzi, S.; Carducci, C.; Dionisi-Vici, C.; Teofoli, F.; Burlina, A.; Angeloni, A.; Aronica, T.; Bordugo, A.; et al. Expanded Newborn Screening in Italy Using Tandem Mass Spectrometry: Two Years of National Experience. IJNS 2022, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Burrage, L.C.; Gibson, J.B.; Strenk, M.E.; Lose, E.J.; Bick, D.P.; Elsea, S.H.; Sutton, V.R.; Sun, Q.; Graham, B.H.; et al. Recurrent ACADVL Molecular Findings in Individuals with a Positive Newborn Screen for Very Long Chain Acyl-coA Dehydrogenase (VLCAD) Deficiency in the United States. Mol. Genet. Metab. 2015, 116, 139–145. [Google Scholar] [CrossRef]
- Sharma, S.; McKenzie, M. The Pathogenesis of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency. Biomolecules 2025, 15, 416. [Google Scholar] [CrossRef]
- Leslie, N.; Saenz-Ayala, S. Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency - GeneReviews®—NCBI Bookshelf. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6816/ (accessed on 23 April 2025).
- Wilcken, B. Fatty Acid Oxidation Disorders: Outcome and Long-term Prognosis. J. Inher. Metab. Disea. 2010, 33, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, L.; Haussmann, U.; Mueller, M.; Spiekerkoetter, U. VLCAD Enzyme Activity Determinations in Newborns Identified by Screening: A Valuable Tool for Risk Assessment. J. Inher. Metab. Disea. 2012, 35, 269–277. [Google Scholar] [CrossRef]
- Flowers, M.; Dickson, A.; Miller, M.J.; Spector, E.; Enns, G.M.; Baudet, H.; Pasquali, M.; Racacho, L.; Sadre-Bazzaz, K.; Wen, T.; et al. Specifications of the ACMG/AMP Guidelines for ACADVL Variant Interpretation. Mol. Genet. Metab. 2023, 140, 107668. [Google Scholar] [CrossRef]
- Hesse, J.; Braun, C.; Behringer, S.; Matysiak, U.; Spiekerkoetter, U.; Tucci, S. The Diagnostic Challenge in Very-long Chain acyl-CoA Dehydrogenase Deficiency (VLCADD). J. Inher. Metab. Disea. 2018, 41, 1169–1178. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucleic Acids Res. 2014, 42, D980-985. [Google Scholar] [CrossRef]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD® ): 2003 Update: HGMD 2003 UPDATE. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, T.; Takagi, T. Estimating Transcription Factor Bindability on DNA. Bioinformatics 1999, 15, 622–630. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neubock, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef]
- Piva, F.; Giulietti, M.; Nocchi, L.; Principato, G. SpliceAid: A Database of Experimental RNA Target Motifs Bound by Splicing Proteins in Humans. Bioinformatics 2009, 25, 1211–1213. [Google Scholar] [CrossRef]
- The Galaxy Community; Abueg, L.A.L.; Afgan, E.; Allart, O.; Awan, A.H.; Bacon, W.A.; Baker, D.; Bassetti, M.; Batut, B.; Bernt, M.; et al. The Galaxy Platform for Accessible, Reproducible, and Collaborative Data Analyses: 2024 Update. Nucleic Acids Res. 2024, 52, W83–W94. [Google Scholar] [CrossRef]
- Ferrie, J.J.; Karr, J.P.; Graham, T.G.W.; Dailey, G.M.; Zhang, G.; Tjian, R.; Darzacq, X. P300 Is an Obligate Integrator of Combinatorial Transcription Factor Inputs. Mol. Cell 2024, 84, 234–243.e4. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.C.; Katneni, U.; Jankowska, K.I.; Meyer, D.; Kimchi-Sarfaty, C. In Silico Methods for Predicting Functional Synonymous Variants. Genome Biol. 2023, 24, 126. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. miRDB: An Online Database for Prediction of Functional microRNA Targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Yuan, H.; Liu, Z.; Chen, M.; Xu, Q.; Jiang, Y.; Zhang, T.; Suo, C.; Chen, X. Protein Truncating Variants in Mitochondrial-Related Nuclear Genes and the Risk of Chronic Liver Disease. BMC Med. 2024, 22, 239. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.T.; Uddin, M.; De Rubeis, S.; Chan, Y.; Kamumbu, A.S.; Zhang, X.; D’Gama, A.M.; Kim, S.N.; Hill, R.S.; Goldberg, A.P.; et al. Rates, Distribution and Implications of Postzygotic Mosaic Mutations in Autism Spectrum Disorder. Nat. Neurosci. 2017, 20, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Andresen, B.S.; Nation, J.; Boneh, A. VLCAD Deficiency: Follow-up and Outcome of Patients Diagnosed through Newborn Screening in Victoria. Mol. Genet Metab. 2016, 118, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Men, S.; Liu, S.; Zheng, Q.; Yang, S.; Mao, H.; Wang, Z.; Gu, Y.; Tang, X.; Wang, L. Incidence and Genetic Variants of Inborn Errors of Metabolism Identified through Newborn Screening: A 7-Year Study in Eastern Coastal Areas of China. Mol. Genet Genomic Med. 2023, 11, e2152. [Google Scholar] [CrossRef]
- Pena, L.D.M.; Van Calcar, S.C.; Hansen, J.; Edick, M.J.; Walsh Vockley, C.; Leslie, N.; Cameron, C.; Mohsen, A.-W.; Berry, S.A.; Arnold, G.L.; et al. Outcomes and Genotype-Phenotype Correlations in 52 Individuals with VLCAD Deficiency Diagnosed by NBS and Enrolled in the IBEM-IS Database. Mol. Genet. Metab. 2016, 118, 272–281. [Google Scholar] [CrossRef]
- Andresen, B.S.; Olpin, S.; Poorthuis, B.J.H.M.; Scholte, H.R.; Vianey-Saban, C.; Wanders, R.; Ijlst, L.; Morris, A.; Pourfarzam, M.; Bartlett, K.; et al. Clear Correlation of Genotype with Disease Phenotype in Very–Long-Chain Acyl-CoA Dehydrogenase Deficiency. Am. J. Hum. Genet. 1999, 64, 479–494. [Google Scholar] [CrossRef]
- Ladd, A.N.; Charlet-B, N.; Cooper, T.A. The CELF Family of RNA Binding Proteins Is Implicated in Cell-Specific and Developmentally Regulated Alternative Splicing. Mol. Cell. Biol. 2001, 21, 1285–1296. [Google Scholar] [CrossRef]
- Young, P.J. SRp30c-Dependent Stimulation of Survival Motor Neuron (SMN) Exon 7 Inclusion Is Facilitated by a Direct Interaction with hTra2beta1. Hum. Mol. Genet. 2002, 11, 577–587. [Google Scholar] [CrossRef]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP Family: Insights into Their Role in Health and Disease. Hum. Genet 2016, 135, 851–867. [Google Scholar] [CrossRef]
- Tummolo, A.; Melpignano, L. The Reciprocal Interplay between Infections and Inherited Metabolic Disorders. Microorganisms 2023, 11, 2545. [Google Scholar] [CrossRef]
- Fatehi, F.; Okhovat, A.A.; Nilipour, Y.; Mroczek, M.; Straub, V.; Töpf, A.; Palibrk, A.; Peric, S.; Rakocevic Stojanovic, V.; Najmabadi, H.; et al. Adult-onset Very-long-chain acyl-CoA Dehydrogenase Deficiency (VLCADD). Euro J. Neurol. 2020, 27, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Van Calcar, S.C.; Sowa, M.; Rohr, F.; Beazer, J.; Setlock, T.; Weihe, T.U.; Pendyal, S.; Wallace, L.S.; Hansen, J.G.; Stembridge, A.; et al. Nutrition Management Guideline for Very-Long Chain Acyl-CoA Dehydrogenase Deficiency (VLCAD): An Evidence- and Consensus-Based Approach. Mol. Genet. Metab. 2020, 131, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Rouyer, A.; Tard, C.; Dessein, A.; Spinazzi, M.; Bédat-Millet, A.; Dimitri-Boulos, D.; Nadaj-Pakleza, A.; Chanson, J.; Nicolas, G.; Douillard, C.; et al. Long-term Prognosis of Fatty-acid Oxidation Disorders in Adults: Optimism despite the Limited Effective Therapies Available. Euro J. Neurol. 2024, 31, e16138. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| NM_000018.3 ACADVL | Position | dbSNP rs | dbSNP Frequency European Population | gnomAD Genomes Frequency European Population | ClinVar Classification | HGMD Classification | Inheritance |
|---|---|---|---|---|---|---|---|
| c.-64T>C | Promoter | rs77051465 | 0.13 | 0.008 | Benign | Not reported | Paternal |
| c.848T>C (p.Val283Ala) | Exon 9 | rs113994167 | 0.002 | 0.001 | Pathogenic | Disease-causing Mutation | Maternal |
| c.957G>A (p.Ser319=) | Exon 10 | rs143870522 | 0.0001 | 0.00003 | Conflicting classifications of pathogenicity | Possible disease-causing mutation (uncertain/less confident) | Paternal |
| NM_000018.3 ACADVL dbSNP rs | Position | gnomAD Total Frequency | ClinVar Classification | HGMD Classification | Supportive Evidence of Pathogenicity | Genotype | Study Population | Ref. |
|---|---|---|---|---|---|---|---|---|
| c.864C>T, p.Phe288= rs753748672 | Exon 9 | 0.00001 | LB | DM? | RA 31% | Homozygous | NBS | [9] |
| c.957G>A, p.Ser319= rs143870522 | Exon 10 | 0.00003 | Conflicting | DM? | RA 24% | Compound heterozygous with c.848T>C, p.Val283Ala, like in this study | NBS | [9] |
| RA 23% | Compound heterozygous with c.1332+2T>A | NBS | [9] | |||||
| c.1077G>A, p.Ala359= rs779458466 | Exon 10 | 0.00001 | Conflicting | DM | Predicted to affect splicing | Homozygous | NBS, symptomatic | [24] |
| Compound heterozygous | NBS | [3] | ||||||
| Heterozygous, de novo | ASD | [23] | ||||||
| c.1317T>A, p.Gly439= rs2142985210 | Exon 13 | 0.000001 | LB | DM | NR | Heterozygous | Individual from the UK Biobank with liver dysfunction | [22] |
| c.1464C>T, p.Gly488= | Exon 15 | - | - | DM? | PM2+PP4 | Compound heterozygous with c.1795G>A, p.E599K | NBS in China, symtomatic | [25] |
| c.1501C>T, p.Leu501 | Exon 15 | - | - | DM | Predicted to affect splicing (missplicing) | Compound heterozygous with c.865G>A, p.G289R | NBS | [26] |
| c.1617T>C, p.Ala539= rs1555528948 | Exon 17 | - | VUS | DM | Predicted to affect splicing by Mutation Taster | Compound heterozygous with c.1708_1717GACGGGGCCA | NBS, symptomatic | [26] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldo, F.; Zupin, L.; Magnolato, A.; Capaci, V.; Bonati, M.T. ACADVL Deep Sequencing in a Case Study: Beyond the Common c.848T>C Pathogenic Variant. Genes 2025, 16, 538. https://doi.org/10.3390/genes16050538
Baldo F, Zupin L, Magnolato A, Capaci V, Bonati MT. ACADVL Deep Sequencing in a Case Study: Beyond the Common c.848T>C Pathogenic Variant. Genes. 2025; 16(5):538. https://doi.org/10.3390/genes16050538
Chicago/Turabian StyleBaldo, Francesco, Luisa Zupin, Andrea Magnolato, Valeria Capaci, and Maria Teresa Bonati. 2025. "ACADVL Deep Sequencing in a Case Study: Beyond the Common c.848T>C Pathogenic Variant" Genes 16, no. 5: 538. https://doi.org/10.3390/genes16050538
APA StyleBaldo, F., Zupin, L., Magnolato, A., Capaci, V., & Bonati, M. T. (2025). ACADVL Deep Sequencing in a Case Study: Beyond the Common c.848T>C Pathogenic Variant. Genes, 16(5), 538. https://doi.org/10.3390/genes16050538