
Competitive Endogenous RNA Network Involving Immune Subgroups, Infiltration, and lncRNAs in Prostate Cancer

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Download
2.2. Immunological Grouping and Its Validation
2.3. Tumor Microenvironment
2.4. Assessment of Immune Cell Infiltration
2.5. Construction of a Weighted Gene Co-Expression Network
2.6. Construction of ceRNA Network
2.7. Functional Enrichment Analysis
2.8. Statistical Analysis
3. Results
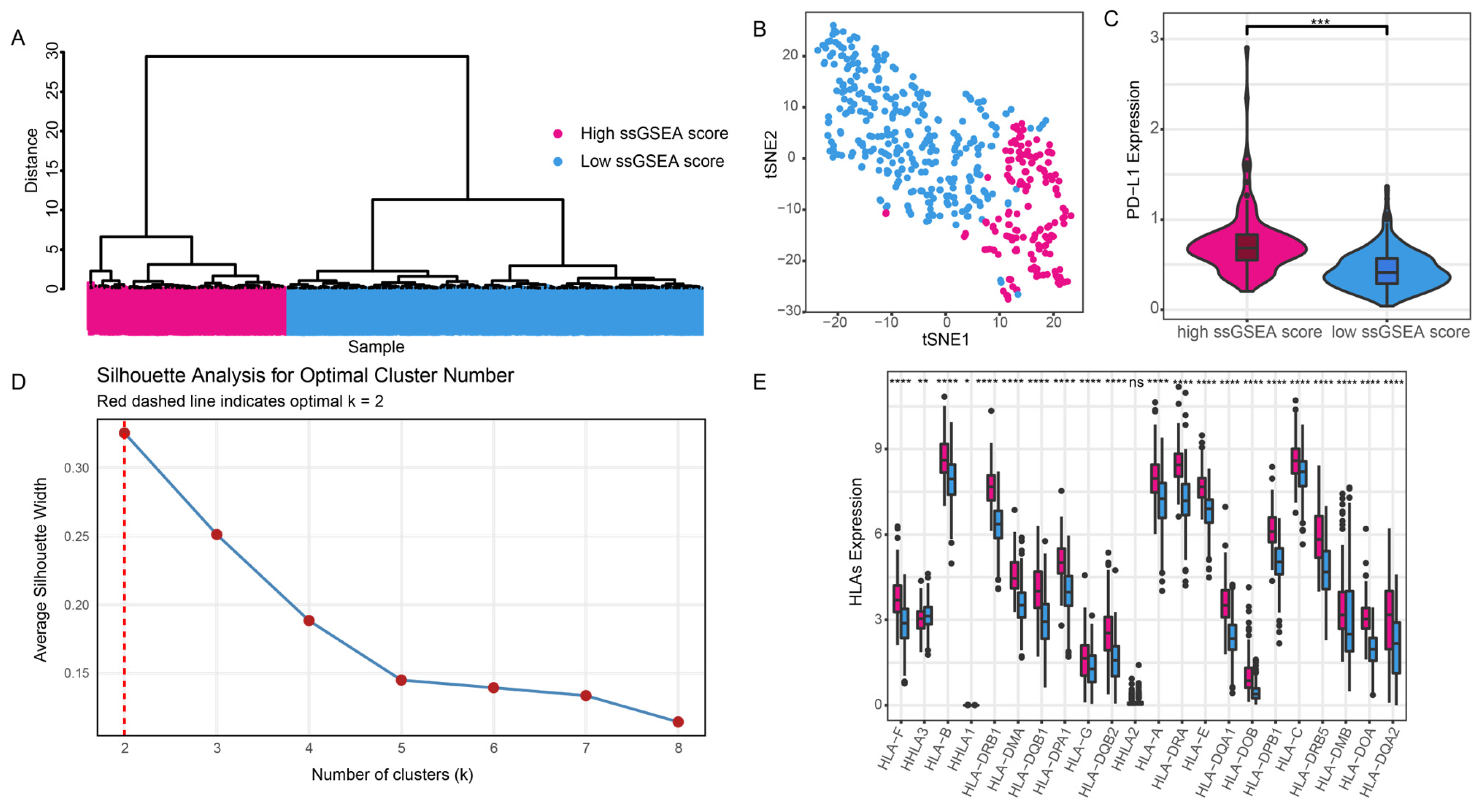
3.1. Immune Grouping
3.2. Differences in Immune Grouping in the Tumor Microenvironment
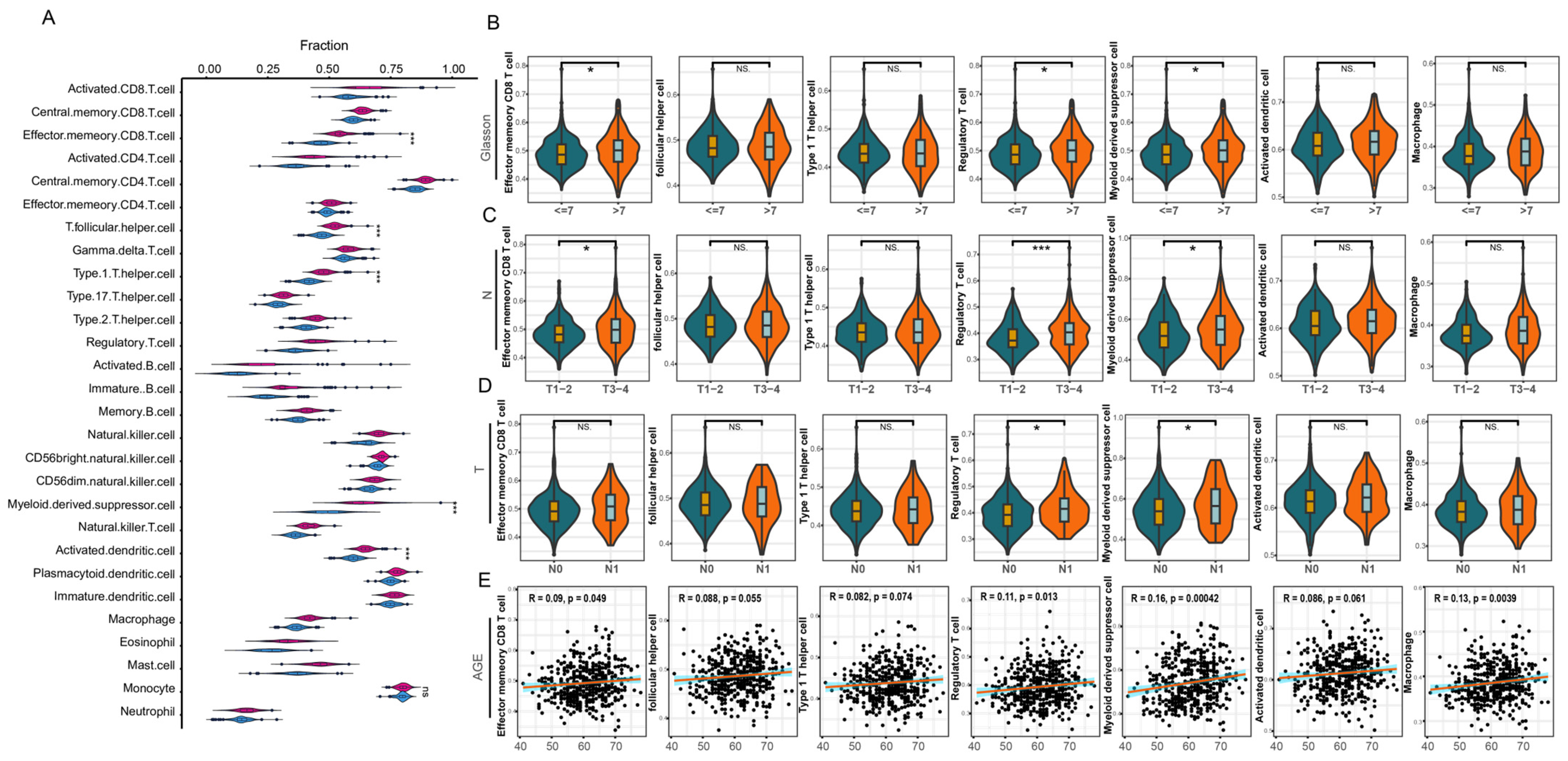
3.3. Infiltration Analysis of Immune Cells
3.4. lncRNAs Associated with the Seven Immune Cells
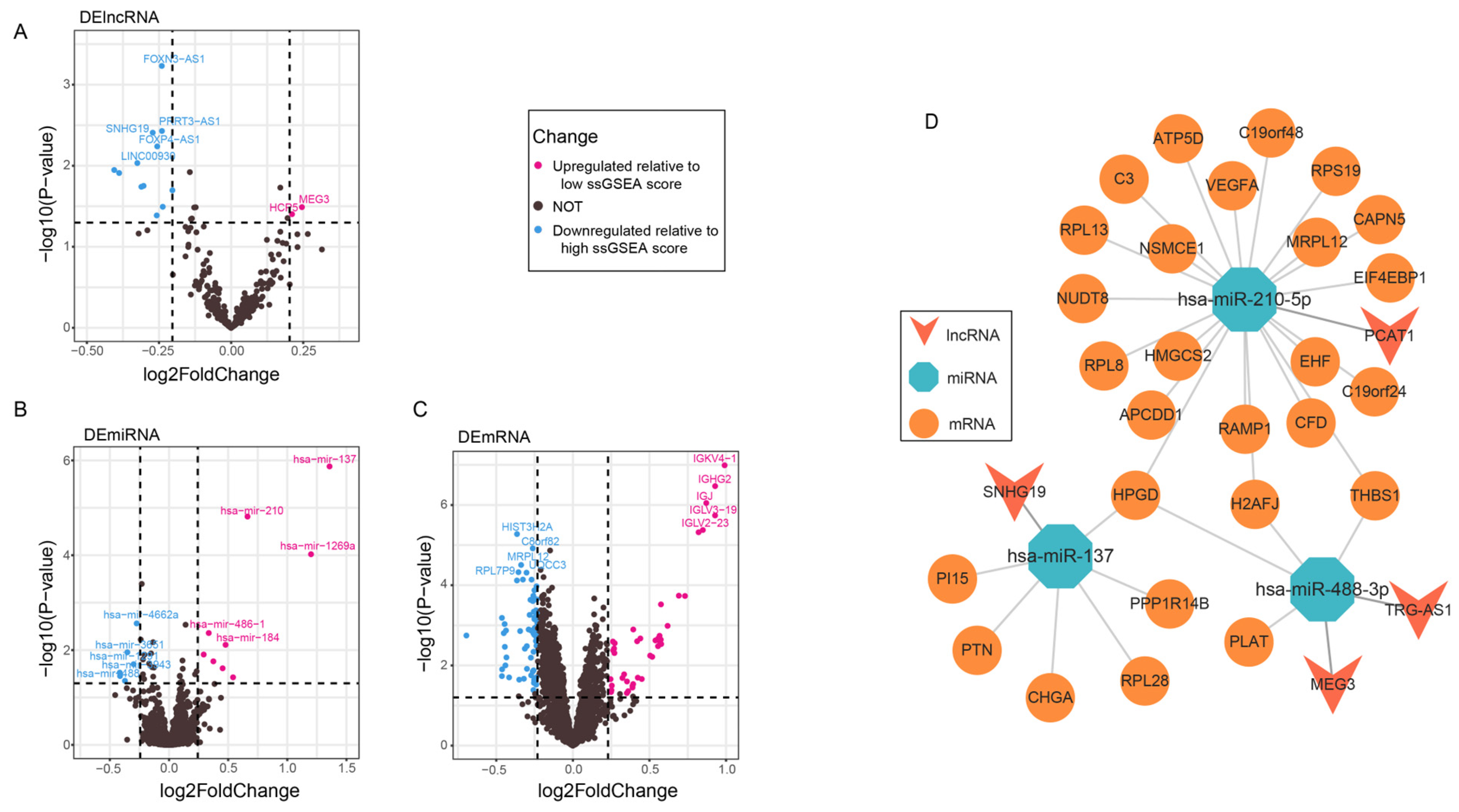
3.5. Construction of the ceRNA Network
3.6. Binding of Seed Regions Between mRNAs, miRNAs, and lncRNAs in PCa
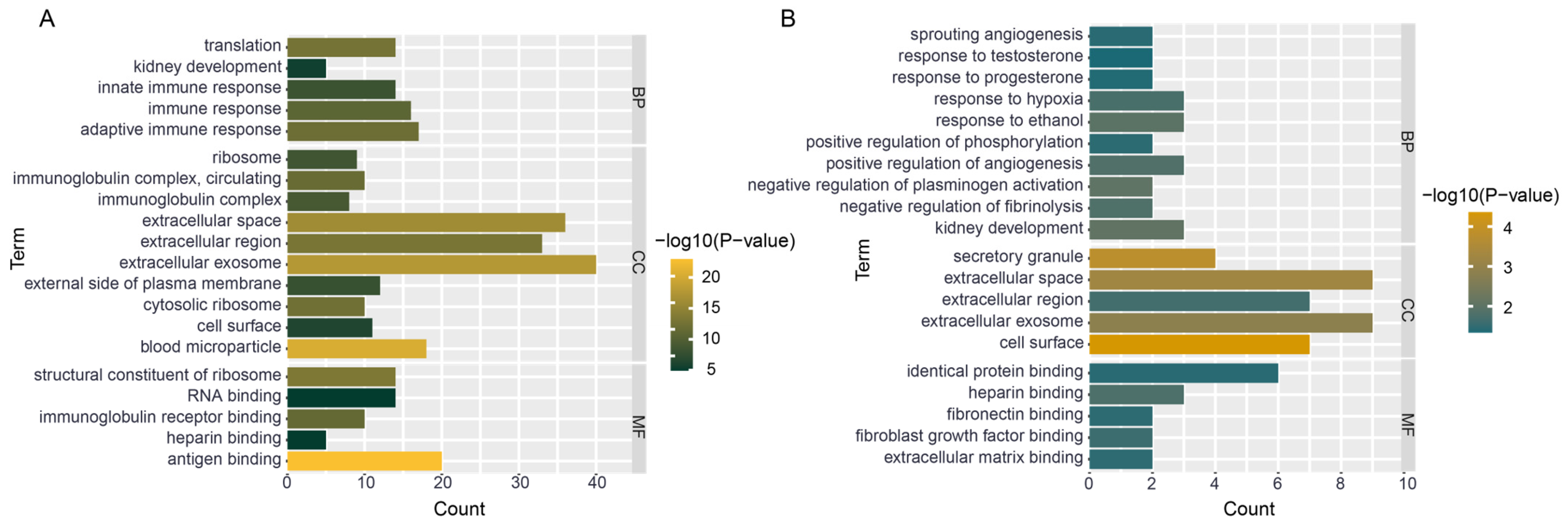
3.7. Functional Enrichment of Hub Genes
4. Discussion
4.1. Immune Infiltrating Cells
4.2. ceRNA Network
4.3. mRNA Functional Enrichment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Ma, J.; Siegel, R.; Fedewa, S.; Brawley, O.; Ward, E.M. Prostate Cancer Incidence Rates 2 Years After the US Preventive Services Task Force Recommendations Against Screening. JAMA Oncol. 2016, 2, 1657–1660. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Rutherford, M.J.; Bardot, A.; Ferlay, J.; Andersson, T.M.; Myklebust, T.Å.; Tervonen, H.; Thursfield, V.; Ransom, D.; Shack, L.; et al. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995–2014 (ICBP SURVMARK-2): A population-based study. Lancet Oncol. 2019, 20, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Terakawa, T.; Jimbo, N.; Inaba, R.; Nakano, Y.; Fujisawa, M. Clinical Features of Treatment-related Neuroendocrine Prostate Cancer: A Case Series. Anticancer Res. 2020, 40, 3519–3526. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Peng, M.; Yi, L. UBASH3B Is a Novel Prognostic Biomarker and Correlated With Immune Infiltrates in Prostate Cancer. Front. Oncol. 2020, 9, 1517. [Google Scholar] [CrossRef]
- Zhou, Y.; Abraham, S.; Andre, P.; Edelstein, L.C.; Shaw, C.A.; Dangelmaier, C.A.; Tsygankov, A.Y.; Kunapuli, S.P.; Bray, P.F.; Mckenzie, S.E. Anti-miR-148a regulates platelet FcγRIIA signaling and decreases thrombosis in vivo in mice. Blood J. Am. Soc. Hematol. 2015, 126, 2871–2881. [Google Scholar] [CrossRef]
- Ge, S.; Mi, Y.; Zhao, X.; Hu, Q.; Guo, Y.; Zhong, F.; Zhang, Y.; Xia, G.; Sun, C. Characterization and validation of long noncoding RNAs as new candidates in prostate cancer. Cancer Cell Int. 2020, 20, 531. [Google Scholar] [CrossRef]
- Wu, H.; Dai, D.; Wang, X. A Novel Radar HRRP Recognition Method with Accelerated T-Distributed Stochastic Neighbor Embedding and Density-Based Clustering. Sensors 2019, 19, 5112. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef]
- Chen, C.; Liu, X.; Chang, C.Y.; Wang, H.Y.; Wang, R.F. The Interplay between T Cells and Cancer: The Basis of Immunotherapy. Genes 2023, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Gollapudi, S. Effector Memory CD8+ T Cells Are Resistant to Apoptosis. Ann. N. Y. Acad. Sci. 2010, 1109, 145–150. [Google Scholar] [CrossRef]
- Ranganathan, R.; Bourdery, L.; Dullaers, M.; Morita, R.; Lavecchio, E.M.; Banchereau, J.; Schmitt, N.; Oh, S.; Punaro, M.; Pascual, V.; et al. Human Blood CXCR5(+)CD4(+) T Cells Are Counterparts of T Follicular Cells and Contain Specific Subsets that Differentially Support Antibody Secretion. Immunity 2011, 34, 108–121. [Google Scholar] [CrossRef]
- Lohning, M.; Hegazy, A.N.; Pinschewer, D.D.; Busse, D.; Lang, K.S.; Hofer, T.; Radbruch, A.; Zinkernagel, R.M.; Hengartner, H. Long-lived virus-reactive memory T cells generated from purified cytokine-secreting T helper type 1 and type 2 effectors. J. Exp. Med. 2008, 205, 53–61. [Google Scholar] [CrossRef]
- Whiteside, T.L. The role of regulatory T cells in cancer immunology. Immunotargets Ther. 2015, 4, 159–171. [Google Scholar] [CrossRef]
- Nair, R.R.; Sinha, P.; Khanna, A.; Singh, K. Reduced Myeloid-derived Suppressor Cells in the Blood and Endometrium is Associated with Early Miscarriage. Am. J. Reprod. Immunol. 2015, 73, 479–486. [Google Scholar] [CrossRef]
- Ma, E.S.; Wang, Z.X.; Zhu, M.Q.; Zhao, J. Immune evasion mechanisms and therapeutic strategies in gastric cancer. World J. Gastrointest. Oncol. 2022, 14, 216–229. [Google Scholar] [CrossRef]
- Guo, Z.H.; Han, L.; Fu, Y.X.; Wu, Z.Y.; Ma, Y.H.; Li, Y.P.; Wang, H.Q.; Jiang, L.; Liang, S.N.; Wang, Z.Z.; et al. Systematic Evaluation of the Diagnostic and Prognostic Significance of Competitive Endogenous RNA Networks in Prostate Cancer. Front. Genet. 2020, 11, 785. [Google Scholar] [CrossRef]
- Li, Q.Y.; Chen, B.L.; Song, G.D.; Zeng, K.; Chen, X.; Miao, J.P.; Yuan, X.L.; Liu, J.H.; Wang, Z.H.; Liu, B. Integrated analysis to identify the AC005154.6/hsa-miR-29c-3p/CCNL2 axis as a novel prognostic biomarker associated with immune infiltration in prostate cancer. Cancer Cell Int. 2022, 22, 17. [Google Scholar] [CrossRef]
- Lin, Y.; Qi, X.; Chen, J.; Shen, B. Multivariate competing endogenous RNA network characterization for cancer microRNA biomarker discovery: A novel bioinformatics model with application to prostate cancer metastasis. Precis. Clin. Med. 2022, 5, 2–12. [Google Scholar] [CrossRef]
- Qiu, L.; Yu, Q.; Zhou, Y.; Zheng, S.; Tao, J.; Jiang, Q.; Yuan, G. Functionally impaired follicular helper T cells induce regulatory B cells and CD14+ human leukocyte antigen-DR- cell differentiation in non-small cell lung cancer. Cancer Sci. 2018, 109, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.W.; Chen, Y.X.; Zhao, Q.; Shi, J.; Yang, W.; Zhu, Z.L.; Yu, W.G.; Guan, J.J.; Song, Y.X.; Wu, H.; et al. Increased circulating Tfh17 and PD-1(+)Tfh cells are associated with autoantibodies in Hashimoto’s thyroiditis. Autoimmunity 2018, 51, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Gerhardt, T.; Winkels, H.; Michel, N.A.; Pramod, A.B.; Ghosheh, Y.; Brunel, S.; Buscher, K.; Miller, J.; McArdle, S.; et al. Pathogenic Autoimmunity in Atherosclerosis Evolves from Initially Protective Apolipoprotein B-100-Reactive CD4(+)T-Regulatory Cells. Circulation 2020, 142, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Ladoire, S.; Martin, F.; Ghiringhelli, F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: The paradox of colorectal cancer. Cancer Immunol. Immunother. 2011, 60, 909–918. [Google Scholar] [CrossRef]
- Onda, M.; Kobayashi, K.; Pastan, I. Depletion of regulatory T cells in tumors with an anti-CD25 immunotoxin induces CD8 T cell-mediated systemic antitumor immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 4575–4582. [Google Scholar] [CrossRef]
- Lee, J.M.; Lee, M.H.; Garon, E.; Goldman, J.W.; Salehi-Rad, R.; Baratelli, F.E.; Schaue, D.; Wang, G.; Rosen, F.; Yanagawa, J.; et al. Phase I Trial of Intratumoral Injection of CCL21 Gene-Modified Dendritic Cells in Lung Cancer Elicits Tumor-Specific Immune Responses and CD8(+) T-cell Infiltration. Clin. Cancer Res. 2017, 23, 4556–4568. [Google Scholar] [CrossRef]
- Cheng, X.Q. A Comprehensive Review of HER2 in Cancer Biology and Therapeutics. Genes 2024, 15, 903. [Google Scholar] [CrossRef]
- Bailey, K.N.; Furman, B.D.; Zeitlin, J.; Kimmerling, K.A.; Wu, C.L.; Guilak, F.; Olson, S.A. Intra-articular depletion of macrophages increases acute synovitis and alters macrophage polarity in the injured mouse knee. Osteoarthr. Cartil. 2020, 28, 626–638. [Google Scholar] [CrossRef]
- Ren, W.; Hou, J.; Yang, C.; Wang, H.; Wu, S.; Wu, Y.; Zhao, X.; Lu, C.J.; Research, C.C. Extracellular vesicles secreted by hypoxia pre-challenged mesenchymal stem cells promote non-small cell lung cancer cell growth and mobility as well as macrophage M2 polarization via miR-21-5p delivery. J. Exp. Clin. Cancer Res. 2019, 38, 14. [Google Scholar] [CrossRef]
- Holly, J.M.P.; Broadhurst, J.; Mansor, R.; Bahl, A.; Perks, C.M. Hyperglycemia Promotes TMPRSS2-ERG Gene Fusion in Prostate Cancer Cells via Upregulating Insulin-Like Growth Factor-Binding Protein-2. Front. Endocrinol. 2017, 8, 305. [Google Scholar] [CrossRef]
- Alabert, C.; Jasencakova, Z.; Groth, A. Chromatin Replication and Histone Dynamics. Adv. Exp. Med. Biol. 2017, 1042, 311–333. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Lin, C.H.; Lin, H.Y.; Kuei, C.H.; Zheng, J.Q.; Wang, Y.H.; Lu, L.S.; Lee, F.P.; Hu, C.J.; Wu, D.; et al. Histone 2A Family Member J Drives Mesenchymal Transition and Temozolomide Resistance in Glioblastoma Multiforme. Cancers 2020, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Ghareeb, W.M.; Lu, X.R.; Huang, Y.; Huang, S.H.; Chi, P. Coexpression network analysis linked H2AFJ to chemoradiation resistance in colorectal cancer. J. Cell. Biochem. 2019, 120, 10351–10362. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, H.; Yamagata, S.; Qian, Z.R.; Kagawa, S.; Sakashita, N. Thrombospondin-1 is highly expressed in desmoplastic components of invasive ductal carcinoma of the breast and associated with lymph node metastasis. J. Med. Investig. 2013, 60, 91–96. [Google Scholar] [CrossRef]
- Jayachandran, A.; Anaka, M.; Prithviraj, P.; Hudson, C.; Behren, A. Thrombospondin 1 promotes an aggressive phenotype through epithelial-to-mesenchymal transition in human melanoma. Oncotarget 2014, 5, 5782–5797. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Huang, T.T.; Li, Y.M.; Qiu, H. Upregulation of THBS1 is Related to Immunity and Chemotherapy Resistance in Gastric Cancer. Int. J. Gen. Med. 2021, 14, 4945–4957. [Google Scholar] [CrossRef]
- Yuan, Q.B.; Chu, H.Y.; Ge, Y.Q.; Ma, G.X.; Du, M.L.; Wang, M.L.; Zhang, Z.D.; Zhang, W. LncRNA PCAT1 and its genetic variant rs1902432 are associated with prostate cancer risk. J. Cancer 2018, 9, 1414–1420. [Google Scholar] [CrossRef]
- Zhao, B.; Hou, X.B.; Zhan, H. Long non-coding RNA PCAT-1 over-expression promotes proliferation and metastasis in non-small cell lung cancer cells. Int. J. Clin. Exp. Med. 2015, 8, 18482–18487. [Google Scholar]
- Liu, L.; Liu, Y.C.; Zhuang, C.L.; Xu, W.; Fu, X.; Lv, Z.J.; Wu, H.W.; Mou, L.S.; Zhao, G.P.; Cai, Z.M.; et al. Inducing cell growth arrest and apoptosis by silencing long non-coding RNA PCAT-1 in human bladder cancer. Tumor Biol. 2015, 36, 7685–7689. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Shao, W.; Shen, N. LncRNA SNHG9 is downregulated in osteoarthritis and inhibits chondrocyte apoptosis by downregulating miR-34a through methylation. BMC Musculoskelet. Disord. 2021, 21, 7. [Google Scholar] [CrossRef]
- Li, X.X.; Wang, L.J.; Hou, J.; Liu, H.Y.; Wang, R.; Wang, C.; Xie, W.H. Identification of Long Noncoding RNAs as Predictors of Survival in Triple-Negative Breast Cancer Based on Network Analysis. Biomed Res. Int. 2020, 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.W.; Hsu, C.L.; Tang, C.W.; Chen, X.J.; Huang, H.C.; Juan, H.F. Multiomics Reveals Ectopic ATP Synthase Blockade Induces Cancer Cell Death via a lncRNA-mediated Phospho-signaling Network. Mol. Cell. Proteom. 2020, 19, 1805–1825. [Google Scholar] [CrossRef]
- Rothzerg, E.; Ho, X.D.; Xu, J.; Wood, D.; Märtson, A.; Kõks, S. Upregulation of 15 Antisense Long Non-Coding RNAs in Osteosarcoma. Genes 2021, 12, 1132. [Google Scholar] [CrossRef]
- Zhu, J.; Dai, H.; Li, X.; Guo, L.; Sun, X.; Zheng, Z.; Xu, C. LncRNA TRG-AS1 inhibits bone metastasis of breast cancer by the miR-877-5p/WISP2 axis. Pathol. Res. Pract. 2023, 243, 154360. [Google Scholar] [CrossRef]
- Li, T.H.; Sun, H.W.; Song, L.J.; Yang, B.; Zhang, P.; Yan, D.M.; Liu, X.Z.; Luo, Y.R. Long non-coding RNA MEG3 regulates autophagy after cerebral ischemia/reperfusion injury. Neural Regen. Res. 2022, 17, 824–831. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, R. Molecular Mechanism of Long Chain Non coding RNA MEG3 Regulating the Development of Parkinson’s Disease by Targeting miR-488-3p. Mod. Med. J. 2021, 49, 769–775. [Google Scholar] [CrossRef]
- Giannakakis, A.; Sandaltzopoulos, R.; Greshock, J.; Liang, S.; Huang, J.; Hasegawa, K.; Li, C.S.; O’Brien-Jenkins, A.; Katsaros, D.; Weber, B.L.; et al. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol. Ther. 2008, 7, 255–264. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Fujiwara, T.; Sato, F.; Shimada, Y.; Tanaka, E.; Sakai, Y.; Shimizu, K.; Tsujimoto, G. MicroRNA-210 Regulates Cancer Cell Proliferation through Targeting Fibroblast Growth Factor Receptor-like 1 (FGFRL1). J. Biol. Chem. 2011, 286, 420–428. [Google Scholar] [CrossRef]
- Radosavljevic, M.; Strac, D.S.; Jancic, J.; Samardzic, J. The Role of Pharmacogenetics in Personalizing the Antidepressant and Anxiolytic Therapy. Genes 2023, 14, 1095. [Google Scholar] [CrossRef]
- Wang, R.L.; Liu, W.F.; Liu, X.P.; Liu, X.X.; Tao, H.; Wu, D.; Zhao, Y.; Zou, L. MicroRNA-210 regulates human trophoblast cell line HTR-8/SVneo function by attenuating Notch1 expression: Implications for the role of microRNA-210 in pre-eclampsia. Mol. Reprod. Dev. 2019, 86, 896–907. [Google Scholar] [CrossRef]
- Li, Y.P.; Jin, L.; Wang, F.; Ren, L.; Pen, R.R.; Bo, G.J.; Wang, L. Epigenetic axis of SNHG19/miR-137/TNFAIP1 modulates amyloid beta peptide 25-35-induced SH-SY5Y cytotoxicity. Epigenomics 2022, 14, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.; Horuz, R.; Celik, M.; Kucukcan, A.J.K.J.U. Is there an association between serum prostate-specific antigen values and serum testosterone levels in healthy men? Korean J. Urol. 2014, 55, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Khera, M.; Crawford, D.; Morales, A.; Salonia, A.; Morgentaler, A.J.E.U. A New Era of Testosterone and Prostate Cancer: From Physiology to Clinical Implications. Eur. Urol. 2014, 65, 115–123. [Google Scholar] [CrossRef]
- Boyle, P.; Koechlin, A.; Bota, M.; D’Onofrio, A.; Zaridze, D.G.; Perrin, P.; Fitzpatrick, J.; Burnett, A.L.; Boniol, M.J.B.I. Endogenous and exogenous testosterone and the risk of prostate cancer and increased prostate-specific antigen (PSA) level: A meta-analysis. BJU Int. 2016, 118, 731–741. [Google Scholar] [CrossRef]
- Lydon, J.P.; Demayo, F.J.; Funk, C.R.; Mani, S.K.; Hughes, A.R.; Montgomery, C.A.; Shyamala, G.; Conneely, O.M.; Omalley, B.W. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995, 9, 2266–2278. [Google Scholar] [CrossRef]
- Erzurumlu, Y.; Dogan, H.K.; Catakli, D. Progesterone regulates the endoplasmic reticulum-associated degradation and Unfolded Protein Response axis by mimicking the androgenic stimulation in prostate cancer cells. Mol. Biol. Rep. 2022, 50, 1253–1265. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3′UTR Binding Sites | Pairing With Target Gene | Site Type |
|---|---|---|
| Position 860-866 of THBS1 3′ UTR | 5′GCUGCCUCCCAAAGGAGGGGCAG | 7mer-A1 |
| hsa-miR-210-5p | 3′GUCACACGCCACCCGUCCCCGA | |
| Position 15-21 of HPGD 3′ UTR | 5′NNACAGCUUAUGUGUUAGCCAUAG | 7mer-A1 |
| hsa-miR-210-5p | 3′CUGGUAUGAUGCCUGUCGGUAA | |
| Position 15-21 of H2AFJ 3′ UTR | 5′NNCCCUGACGCCGCCCUCAGGGAG | 7mer-A1 |
| hsa-miR-210-5p | 3′GUCUUGUCCUGUCCAGUCCCG | |
| Position 308-314 of H2AFJ 3′ UTR | 5′GGUCACCCUCGAGGCGUCAGGGG | 7mer-m8 |
| hsa-miR-210-5p | 3′GUCUUGUCCUGUCCAGUCCCG | |
| Position 175412376-175412399 of HPGD 3′ UTR | 5′AACUUUUAACAGUUACAGCAAUAA | 8mer |
| hsa-miR-137 | 3′GAUGCGCAUAAGAAU-UCGUUAUU | |
| Position 175412827-175412834 of HPGD 3′ UTR | 5′AUUCAAUAUUCUGCCUUUCAG | 7mer-m8 |
| hsa-miR-488-3p | 3′CUGGUUCUUUAUCGGAAAGUU | |
| Position 14928678-14928683 of H2AFJ 3′ UTR | 5′UCGGUUUGACUUUGCUUUCAA | 7mer-A1 |
| hsa-miR-488-3p | 3′CUGGUUCUUUAUCGGAAAGUU | |
| Position 14928420-14928425 of H2AFJ 3′ UTR | 5′UAGUAGCUGCUGUGCUUUCAU | 6mer |
| hsa-miR-488-3p | 3′CUGGUUCUUUAUCGGAAAGUU | |
| Position 39889031-39889036 of THBS1 3′ UTR | 5′AUAUACACUUUUUUCUUUCAU | 6mer |
| hsa-miR-488-3p | 3′CUGGUUCUUUAUCGGAAAGUU |
| lncRNA/miRNA | Pairing with Target Gene | Site Type |
|---|---|---|
| MEG3 | Target: 5′agucucugucuuuCCUUUCAc 3′ | 7mer-m8 |
| hsa-miR-488-3p | miRNA: 3′cugguucuuuaucGGAAAGUu 5′ | |
| TRG-AS1 | Target: 5′aaguguGAAUUA-CCUUUCAu 3′ | 7mer-m8 |
| hsa-miR-488-3p | miRNA: 3′cugguuCUUUAUCGGAAAGUu 5′ | |
| SNHG19 | Target: 5′cuauugaaGUGCUU-AGCAAUAa 3′ | 8mer |
| hsa-miR-137 | miRNA: 3′gaugcgcaUAAGAAUUCGUUAUu 5′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, W.; Zhang, T.; Ma, L. Competitive Endogenous RNA Network Involving Immune Subgroups, Infiltration, and lncRNAs in Prostate Cancer. Genes 2025, 16, 527. https://doi.org/10.3390/genes16050527
Niu W, Zhang T, Ma L. Competitive Endogenous RNA Network Involving Immune Subgroups, Infiltration, and lncRNAs in Prostate Cancer. Genes. 2025; 16(5):527. https://doi.org/10.3390/genes16050527
Chicago/Turabian StyleNiu, Wenkang, Tingting Zhang, and Lei Ma. 2025. "Competitive Endogenous RNA Network Involving Immune Subgroups, Infiltration, and lncRNAs in Prostate Cancer" Genes 16, no. 5: 527. https://doi.org/10.3390/genes16050527
APA StyleNiu, W., Zhang, T., & Ma, L. (2025). Competitive Endogenous RNA Network Involving Immune Subgroups, Infiltration, and lncRNAs in Prostate Cancer. Genes, 16(5), 527. https://doi.org/10.3390/genes16050527