
A Novel Frameshift Variant and a Partial EHMT1 Microdeletion in Kleefstra Syndrome 1 Patients Resulting in Variable Phenotypic Severity and Literature Review

 ,
, 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Editorial Policies and Ethical Considerations
2.2. DNA Extraction
2.3. Exome Sequencing (ES)
2.4. Chromosomal Microarray Analysis (CMA)
2.5. Sanger Sequencing
3. Case Reports
3.1. Patient 1 (P1)
3.2. Patient 2 (P2)
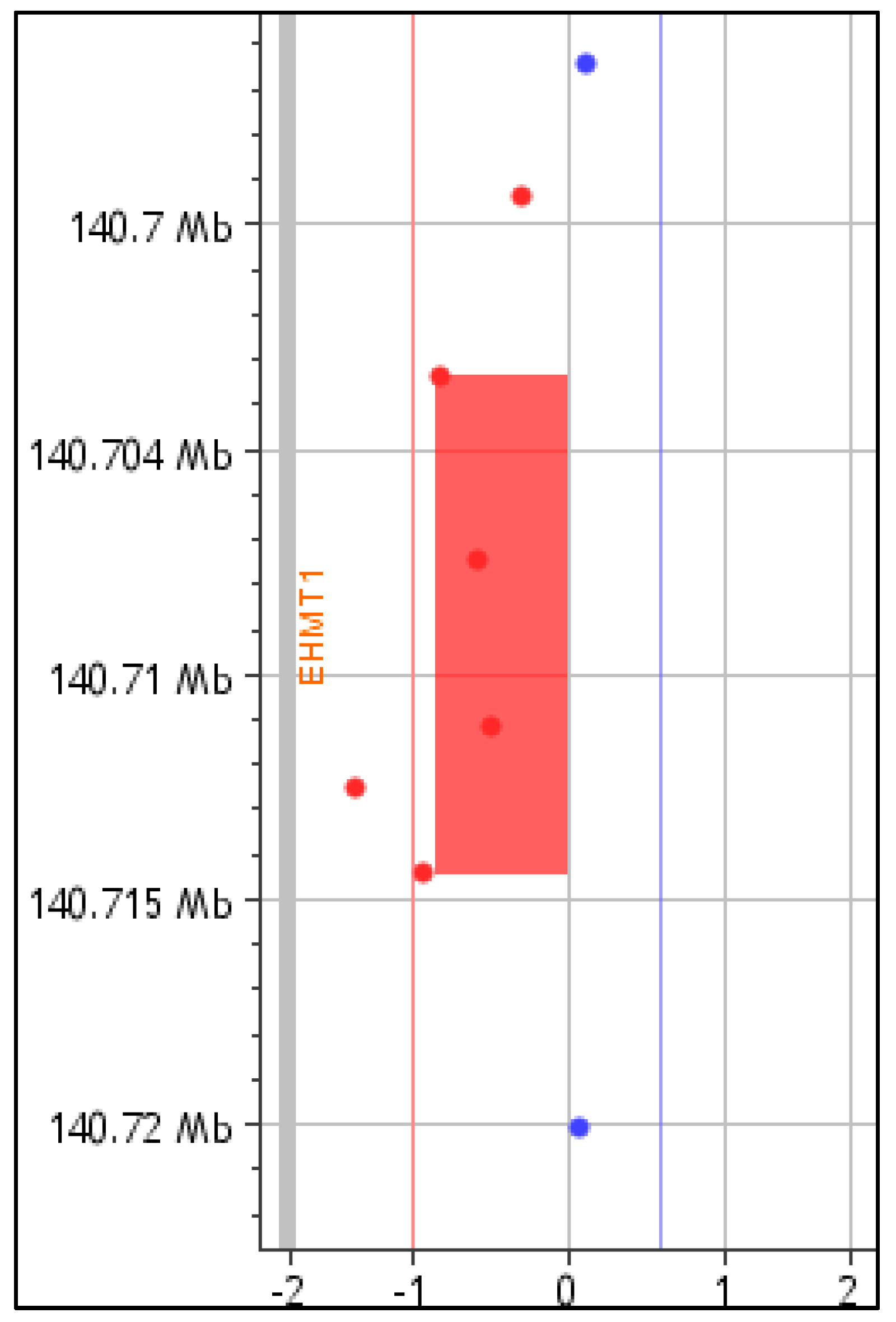
4. Results
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rots, D.; Bouman, A.; Yamada, A.; Levy, M.; Dingemans, A.J.M.; de Vries, B.B.A.; Ruiterkamp-Versteeg, M.; de Leeuw, N.; Ockeloen, C.W.; Pfundt, R.; et al. Comprehensive EHMT1 Variants Analysis Broadens Genotype-Phenotype Associations and Molecular Mechanisms in Kleefstra Syndrome. Am. J. Hum. Genet. 2024, 111, 1605–1625. [Google Scholar] [CrossRef] [PubMed]
- Ciaccio, C.; Scuvera, G.; Tucci, A.; Gentilin, B.; Baccarin, M.; Marchisio, P.; Avignone, S.; Milani, D. New Insights into Kleefstra Syndrome: Report of Two Novel Cases with Previously Unreported Features and Literature Review. Cytogenet. Genome Res. 2019, 156, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Da Cás, E.; Pires, L.V.L.; Linnenkamp, B.D.W.; Allegro, M.C.; Honjo, R.S.; Bertola, D.R.; Aoi, H.; Matsumoto, N.; Kim, C.A. The First Brazilian Clinical Report of Kleefstra Syndrome, Including Semicircular Canals Agenesis as a Possible Phenotype Expansion. Eur. J. Med. Genet. 2024, 71, 104966. [Google Scholar] [CrossRef] [PubMed]
- Frazier, Z.J.; Kilic, S.; Osika, H.; Mo, A.; Quinn, M.; Ballal, S.; Katz, T.; Shearer, A.E.; Horlbeck, M.A.; Pais, L.S.; et al. Novel Phenotypes and Genotype–Phenotype Correlations in a Large Clinical Cohort of Patients With Kleefstra Syndrome. Clin. Genet. 2025. [Google Scholar] [CrossRef]
- Huang, Q.; Xiong, H.; Tao, Z.; Yue, F.F.; Xiao, N. Clinical Phenotypes and Molecular Findings in Ten Chinese Patients with Kleefstra Syndrome Type 1 Due to EHMT1 Defects. Eur. J. Med. Genet. 2021, 64, 104289. [Google Scholar] [CrossRef]
- Kleefstra, T.; Kramer, J.M.; Neveling, K.; Willemsen, M.H.; Koemans, T.S.; Vissers, L.E.L.M.; Wissink-Lindhout, W.; Fenckova, M.; Van Den Akker, W.M.R.; Kasri, N.N.; et al. Disruption of an EHMT1-Associated Chromatin-Modification Module Causes Intellectual Disability. Am. J. Hum. Genet. 2012, 91, 73–82. [Google Scholar] [CrossRef]
- Kohli, U. Shone’s Complex in a Patient with Chromosome 9q34.3 Deletion (Kleefstra Syndrome). Cardiol. Young 2019, 29, 249–251. [Google Scholar] [CrossRef]
- Kukla, M.; Karoń, M.; Chrościńska-Krawczyk, M. Kleefstra Syndrome—Case Report. Child Neurol. 2019, 28, 49–52. [Google Scholar] [CrossRef]
- Lee, R.; Lee, M.S.; Moon, J.E. A Korean Male with Kleefstra Syndrome Presented with Micropenis. Ann. Pediatr. Endocrinol. Metab. 2023, 28, 308–311. [Google Scholar] [CrossRef]
- Ren, R.; Liu, Y.; Liu, P.; Zhao, J.; Hou, M.; Li, S.; Chen, Z.; Yuan, A. Clinical Characteristics and Genetic Analysis of Four Pediatric Patients with Kleefstra Syndrome. BMC Med. Genom. 2024, 17, 290. [Google Scholar] [CrossRef]
- Willemsen, M.H.; Vulto-Van Silfhout, A.T.; Nillesen, W.M.; Wissink-Lindhout, W.M.; Van Bokhoven, H.; Philip, N.; Berry-Kravis, E.M.; Kini, U.; Van Ravenswaaij-Arts, C.M.A.; Delle Chiaie, B.; et al. Update on Kleefstra Syndrome. Mol. Syndr. 2011, 2, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Kleefstra, T.; Brunner, H.G.; Amiel, J.; Oudakker, A.R.; Nillesen, W.M.; Magee, A.; Geneviève, D.; Cormier-Daire, V.; van Esch, H.; Fryns, J.-P.; et al. Loss-of-Function Mutations in Euchromatin Histone Methyl Transferase 1 (EHMT1) Cause the 9q34 Subtelomeric Deletion Syndrome. Am. J. Hum. Genet. 2006, 79, 370–377. [Google Scholar] [CrossRef]
- Verhoeven, W.M.A.; Egger, J.I.M.; Vermeulen, K.; van de Warrenburg, B.P.C.; Kleefstra, T. Kleefstra Syndrome in Three Adult Patients: Further Delineation of the Behavioral and Neurological Phenotype Shows Aspects of a Neurodegenerative Course. Am. J. Med. Genet. A 2011, 155, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Müller, D.J.; Desarkar, P. Psychiatric Manifestations of Kleefstra Syndrome: A Case Report. Front. Psychiatry 2023, 14, 1174195. [Google Scholar] [CrossRef]
- Niu, M.; Li, Y.; Zhan, S.; Sun, B.; Liu, J.; Wu, Y. Tourette-like Syndrome Secondary to Kleefstra Syndrome 1 with a de Novo Microdeletion in the EHMT1 Gene. BMC Neurol. 2023, 23, 365. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Nag, H.E.; Hunn, B.S.; Houge, G.; Hoxmark, L.B. A Structured Assessment of Motor Function and Behavior in Patients with Kleefstra Syndrome. Eur. J. Med. Genet. 2016, 59, 240–248. [Google Scholar] [CrossRef]
- Frega, M.; Linda, K.; Keller, J.M.; Gümüş-Akay, G.; Mossink, B.; van Rhijn, J.R.; Negwer, M.; Klein Gunnewiek, T.; Foreman, K.; Kompier, N.; et al. Neuronal Network Dysfunction in a Model for Kleefstra Syndrome Mediated by Enhanced NMDAR Signaling. Nat. Commun. 2019, 10, 4928. [Google Scholar] [CrossRef]
- Koemans, T.S.; Kleefstra, T.; Chubak, M.C.; Stone, M.H.; Reijnders, M.R.F.; de Munnik, S.; Willemsen, M.H.; Fenckova, M.; Stumpel, C.T.R.M.; Bok, L.A.; et al. Functional Convergence of Histone Methyltransferases EHMT1 and KMT2C Involved in Intellectual Disability and Autism Spectrum Disorder. PLoS Genet. 2017, 13, e1006864. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an Enhanced Web Interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef]
- Kerchner, K.M.; Mou, T.C.; Sun, Y.; Rusnac, D.V.; Sprang, S.R.; Briknarová, K. The Structure of the Cysteine-Rich Region from Human Histone-Lysine N-Methyltransferase EHMT2 (G9a). J. Struct. Biol. X 2021, 5, 100050. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Ortega, L.; Megías-Fernández, C.; Domínguez-Giménez, P.; Jimeno-González, S.; Rivero, S. Cell Consequences of Loss of Function of the Epigenetic Factor EHMT1. Cell Signal 2023, 108, 110734. [Google Scholar] [CrossRef] [PubMed]
- Alsaqati, M.; Davis, B.A.; Wood, J.; Jones, M.M.; Jones, L.; Westwood, A.; Petter, O.; Isles, A.R.; Linden, D.; Van den Bree, M.; et al. NRSF/REST Lies at the Intersection between Epigenetic Regulation, MiRNA-Mediated Gene Control and Neurodevelopmental Pathways Associated with Intellectual Disability (ID) and Schizophrenia. Transl. Psychiatry 2022, 12, 438. [Google Scholar] [CrossRef]
- Vasireddi, S.K.; Draksler, T.Z.; Bouman, A.; Kummeling, J.; Wheeler, M.; Reuter, C.; Srivastava, S.; Harris, J.; Fisher, P.G.; Narayan, S.M.; et al. Arrhythmias Including Atrial Fibrillation and Congenital Heart Disease in Kleefstra Syndrome: A Possible Epigenetic Link. Europace 2024, 26, euae003. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Yan, L.; Ding, L.; Zhang, Y.; Wang, M.; Yang, Y.; Wu, J.; Chen, C.; Tang, M.; Li, H. Comparison of Genetic, Auditory Features, and Systemic Clinical Phenotype in 14 Families with Syndromic Hearing Loss. Appl. Clin. Genet. 2024, 17, 171–186. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzetis, M.; Mitrakos, A.; Papathanasiou, I.; Koute, V.; Kosma, K.; Pons, R.; Michoula, A.; Grivea, I.; Tsezou, A. A Novel Frameshift Variant and a Partial EHMT1 Microdeletion in Kleefstra Syndrome 1 Patients Resulting in Variable Phenotypic Severity and Literature Review. Genes 2025, 16, 521. https://doi.org/10.3390/genes16050521
Tzetis M, Mitrakos A, Papathanasiou I, Koute V, Kosma K, Pons R, Michoula A, Grivea I, Tsezou A. A Novel Frameshift Variant and a Partial EHMT1 Microdeletion in Kleefstra Syndrome 1 Patients Resulting in Variable Phenotypic Severity and Literature Review. Genes. 2025; 16(5):521. https://doi.org/10.3390/genes16050521
Chicago/Turabian StyleTzetis, Maria, Anastasios Mitrakos, Ioanna Papathanasiou, Vasiliki Koute, Konstantina Kosma, Roser Pons, Aspasia Michoula, Ioanna Grivea, and Aspasia Tsezou. 2025. "A Novel Frameshift Variant and a Partial EHMT1 Microdeletion in Kleefstra Syndrome 1 Patients Resulting in Variable Phenotypic Severity and Literature Review" Genes 16, no. 5: 521. https://doi.org/10.3390/genes16050521
APA StyleTzetis, M., Mitrakos, A., Papathanasiou, I., Koute, V., Kosma, K., Pons, R., Michoula, A., Grivea, I., & Tsezou, A. (2025). A Novel Frameshift Variant and a Partial EHMT1 Microdeletion in Kleefstra Syndrome 1 Patients Resulting in Variable Phenotypic Severity and Literature Review. Genes, 16(5), 521. https://doi.org/10.3390/genes16050521