
An Upper Bound on the Power of DNA to Distinguish Pedigree Relationships

Abstract
1. Introduction
2. Materials and Methods
2.1. Pedigree Relationships and IBD
- Linear relationships: These are relationships between a person and their parent, grandparent (abbreviated GP), great-grandparent (abbreviated GGP), and so on. Beyond the GGP, these relationships are abbreviated as GP with . The GP relationship is referred to as a linear relationship of degree 2, so GP is a linear relationship of degree .
- Cousin relationships: These are relationships between linear descendants of two full siblings. First cousins are labelled 1C, second cousins 2C, and so on. Removal is indicated by the number and the letter R such that first cousins once removed becomes 1C1R. The full sibling relationship could be considered 0th cousins but this relationship is excluded from this study to simplify the analysis by considering the IBD status to be binary (0 or 1).
- Avuncular relationships: These are relationships between a person and (descendants of) a child of their full sibling. The uncle–nephew relationship is abbreviated as N. Great-nephew is abbreviated as GN. For , this relationship is abbreviated as N.
- Half-cousin relationships: These are linear descendants of two half-siblings. These relationships are prefixed with the letter H, for example, H1C2R stands for half first cousins twice removed.
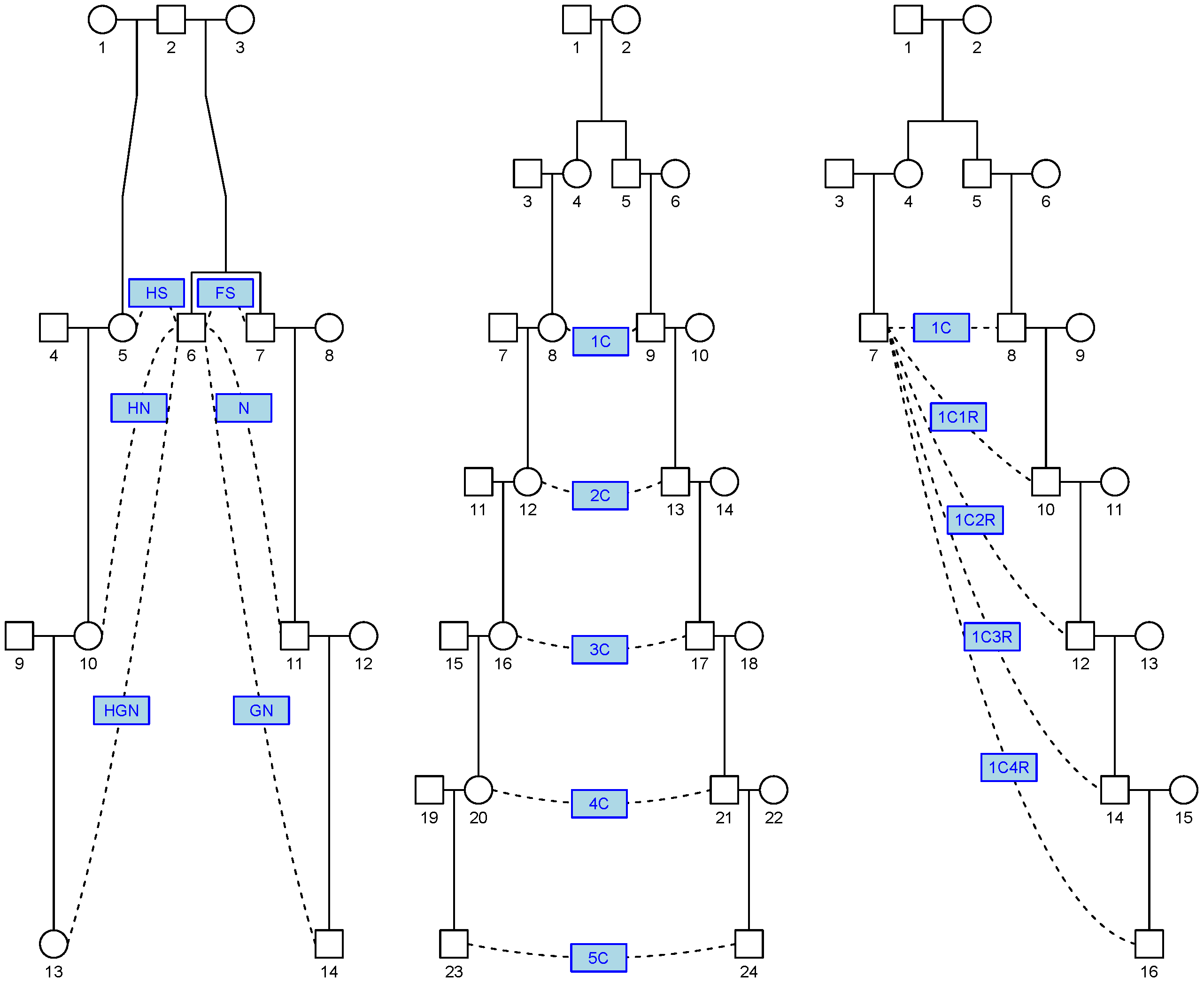
2.2. Relatedness Inference
2.3. Continuous IBD
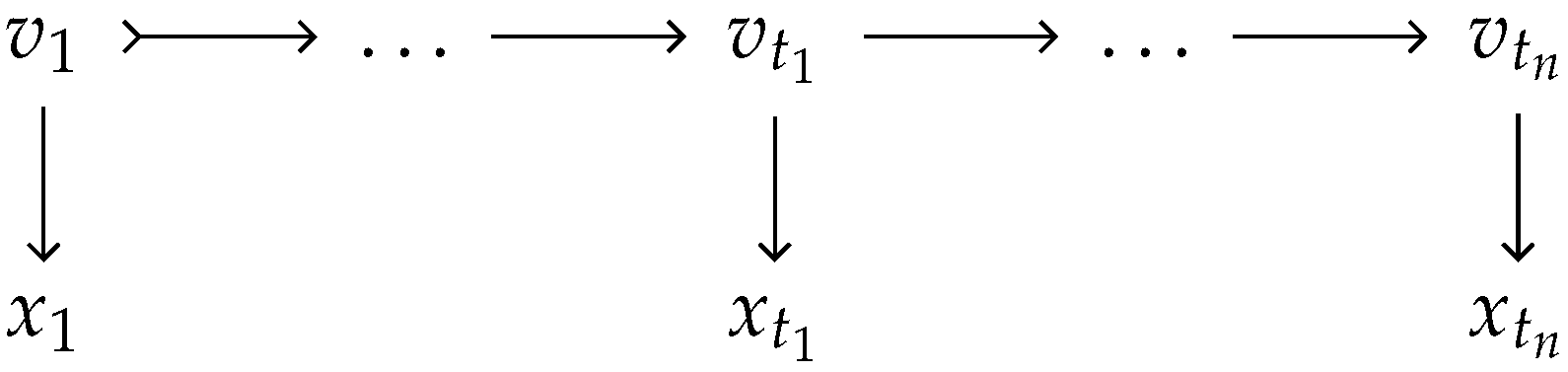
2.3.1. IBD Vector and the Hidden Markov Model
2.3.2. Simulating Continuous IBD
2.3.3. Expectation and Variance of Total IBD
2.3.4. Probability of No IBD:
- since the number of recombination events in the pedigree is Poisson-distributed, , with , where is the number of non-founders in the pedigree.
- The sum over k can be truncated after a finite value . Choosing the -quantile of the Poisson distribution ensures the truncation error is smaller than .
- is the set of paths for which the IBD state remains 0.
- denotes the prior probability of starting in . This probability equals where is the number of IBD vectors.
- is shorthand for .
2.3.5. Likelihoods for IBD Segments
2.3.6. Likelihood Ratios for IBD Segments
2.4. Exploring IBD and Segment Count Distributions
- : the probability of (single) IBD at any point.
- The expectation and standard deviation of (total IBD), the combined length (cM) of all IBD segments.
- The expectation and standard deviation of the segment count.
- ): the probability of not sharing any autosomal DNA across the 22 chromosomes.
- The expectation and standard deviation of , the number of chromosomes for which there is no IBD.
- Segment count: the number of IBD segments across the autosomal genome.
- (total IBD): the combined length (cM) of all IBD segments.
2.5. Empirical LRs for Distinguishing Relationships Using Continuous IBD
- Linear relationships;
- Cousin-type relationships.
3. Results
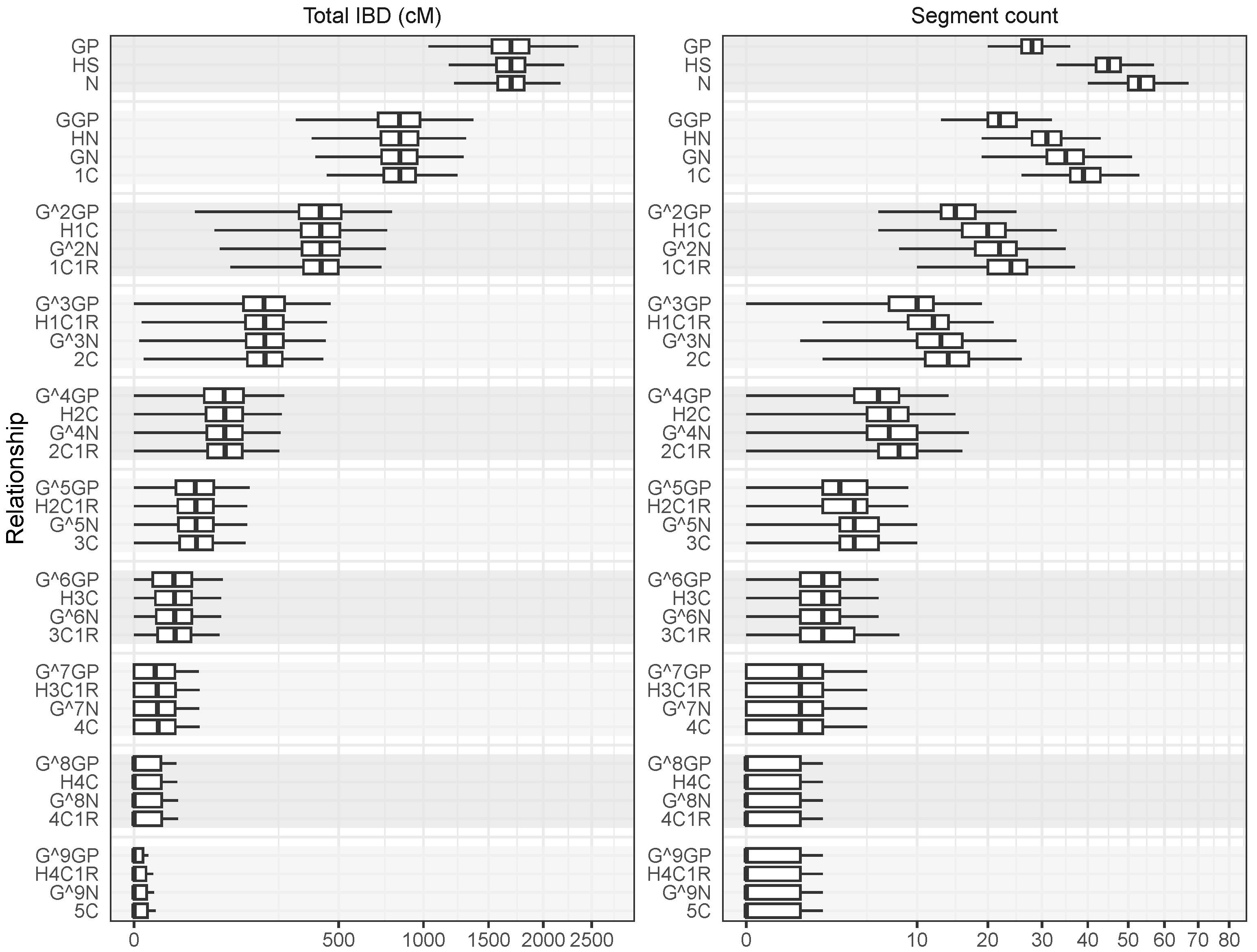
3.1. Exploring Total IBD and Segment Count
- Close relationships (larger ) could be reliably distinguished from distant relationships (smaller ) because the distributions of total IBD were well separated.
- As the relationships became more distant, the total expected IBD decreased while the standard deviation increased relative to the expected value. This means that the distributions of total IBD had more overlap as relationships became more distant.
- Relationships with identical had the same expected total IBD. For example, relationships with included GP, N, and HS, and these were all expected to share 1696 cM. However, their IBD distributions were not the same as evidenced by differences in the standard deviations. Thus, it may be possible to distinguish these relationships based on data beyond total IBD, such as segment count.
- The differences in IBD distributions between relationships with the same decreased quickly as relationships became more distant. The standard deviation of total IBD, Pr(total IBD = 0) and the expected value and standard deviation of the number of chromosomes without IBD all showed a similar trend of convergence within the groups of relationships with the same . It was therefore not possible to reliably distinguish higher-order relationships with the same .
- Many cousin-type relationships such as 2C and 1C2R could not be distinguished because the IBD distributions were identical. Donnelly [19] considered cousin-type relationships of the type “sth cousins t times removed” where and and showed that the IBD distribution depended on .
- For higher-order relationships, there was a substantial probability that no DNA was shared at all. For fifth cousins, there was about a 69% probability of not sharing DNA. For fourth cousins, this probability was about 30%.
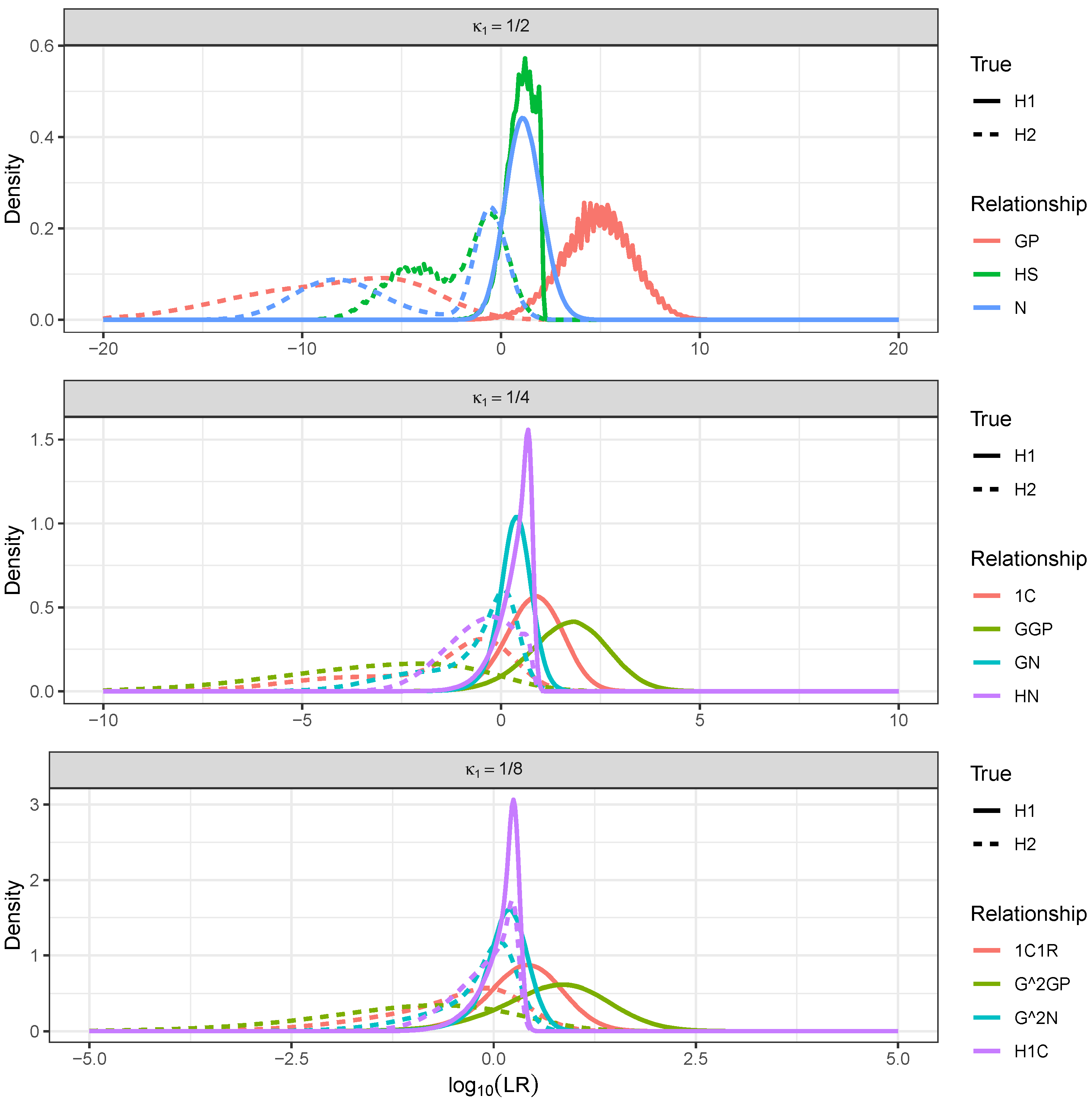
- GP, HS, and N (all with ) had mostly overlapping total IBD distributions but could be distinguished based on segment count.
- GGP, HN, GN, and 1C (all with ) had mostly overlapping total IBD distributions and partly overlapping distributions of segment count.
- There appeared to be limited information in the segment count for distinguishing relationships with smaller than, say, .
- As relationships become more distant, both the distributions of total IBD and segment count became highly skewed.Table 2. IBD distributions summarised for common pedigree relationships.
Relationship Total IBD (cM) Segment Count Expected S.d. Expected S.d. Expected S.d. GP 1/2 1695.68 243.48 27.96 3.38 4.455 × 10−22 2.67 1.51 HS 1/2 1695.68 188.64 44.91 4.44 8.326 × 10−37 0.80 0.86 N 1/2 1695.68 174.35 53.39 4.94 2.091 × 10−43 0.49 0.68 GGP 1/4 847.84 196.31 22.46 4.02 2.264 × 10−12 6.80 2.13 HN 1/4 847.84 173.26 30.94 4.99 1.512 × 10−15 5.03 1.93 GN 1/4 847.84 167.35 35.17 5.56 1.828 × 10−16 4.61 1.87 1C 1/4 847.84 148.60 39.41 5.24 1.148 × 10−21 2.95 1.55 GP 1/8 423.92 139.15 15.47 3.89 1.903 × 10−7 11.07 2.31 H1C 1/8 423.92 127.39 19.71 4.61 9.641 × 10−9 9.74 2.29 N 1/8 423.92 124.22 21.83 5.05 3.854 × 10−9 9.37 2.27 1C1R 1/8 423.92 115.34 23.95 5.02 1.425 × 10−10 8.16 2.22 GP 1/16 211.96 94.45 9.85 3.36 0.0001 14.66 2.18 H1C1R 1/16 211.96 87.97 11.97 3.86 2.860 × 10−5 13.79 2.24 N 1/16 211.96 86.14 13.03 4.16 1.808 × 10−5 13.52 2.25 2C 1/16 211.96 81.46 14.09 4.19 4.761 × 10−6 12.76 2.28 GP 1/32 105.98 63.05 5.99 2.72 0.0050 17.33 1.90 H2C 1/32 105.98 59.33 7.05 3.05 0.0025 16.80 1.97 N 1/32 105.98 58.22 7.58 3.25 0.0019 16.62 2.00 2C1R 1/32 105.98 55.62 8.11 3.29 0.0011 16.18 2.05 GP 1/64 52.99 41.86 3.52 2.12 0.0460 19.14 1.57 H2C1R 1/64 52.99 39.66 4.05 2.33 0.0322 18.84 1.64 N 1/64 52.99 38.98 4.32 2.46 0.0283 18.73 1.66 3C 1/64 52.99 37.49 4.58 2.49 0.0211 18.49 1.71 GP 1/128 26.49 27.80 2.03 1.61 0.1702 20.30 1.25 H3C 1/128 26.49 26.47 2.29 1.75 0.1415 20.14 1.30 N 1/128 26.49 26.04 2.42 1.83 0.1321 20.07 1.32 3C1R 1/128 26.49 25.17 2.56 1.86 0.1144 19.94 1.36 GP 1/256 13.25 18.51 1.15 1.21 0.3653 21.02 0.97 H3C1R 1/256 13.25 17.70 1.28 1.30 0.3319 20.93 1.01 N 1/256 13.25 17.42 1.34 1.35 0.3200 20.89 1.02 4C 1/256 13.25 16.90 1.41 1.37 0.2979 20.82 1.05 GP 1/512 6.62 12.37 0.64 0.89 0.5674 21.44 0.74 H4C 1/512 6.62 11.87 0.71 0.95 0.5400 21.39 0.77 N 1/512 6.62 11.69 0.74 0.99 0.5296 21.37 0.78 4C1R 1/512 6.62 11.38 0.77 1.00 0.5110 21.34 0.80 GP 1/1024 3.31 8.30 0.35 0.66 0.7293 21.69 0.56 H4C1R 1/1024 3.31 7.99 0.39 0.70 0.7109 21.66 0.58 N 1/1024 3.31 7.87 0.40 0.72 0.7037 21.65 0.59 5C 1/1024 3.31 7.68 0.42 0.73 0.6912 21.63 0.60

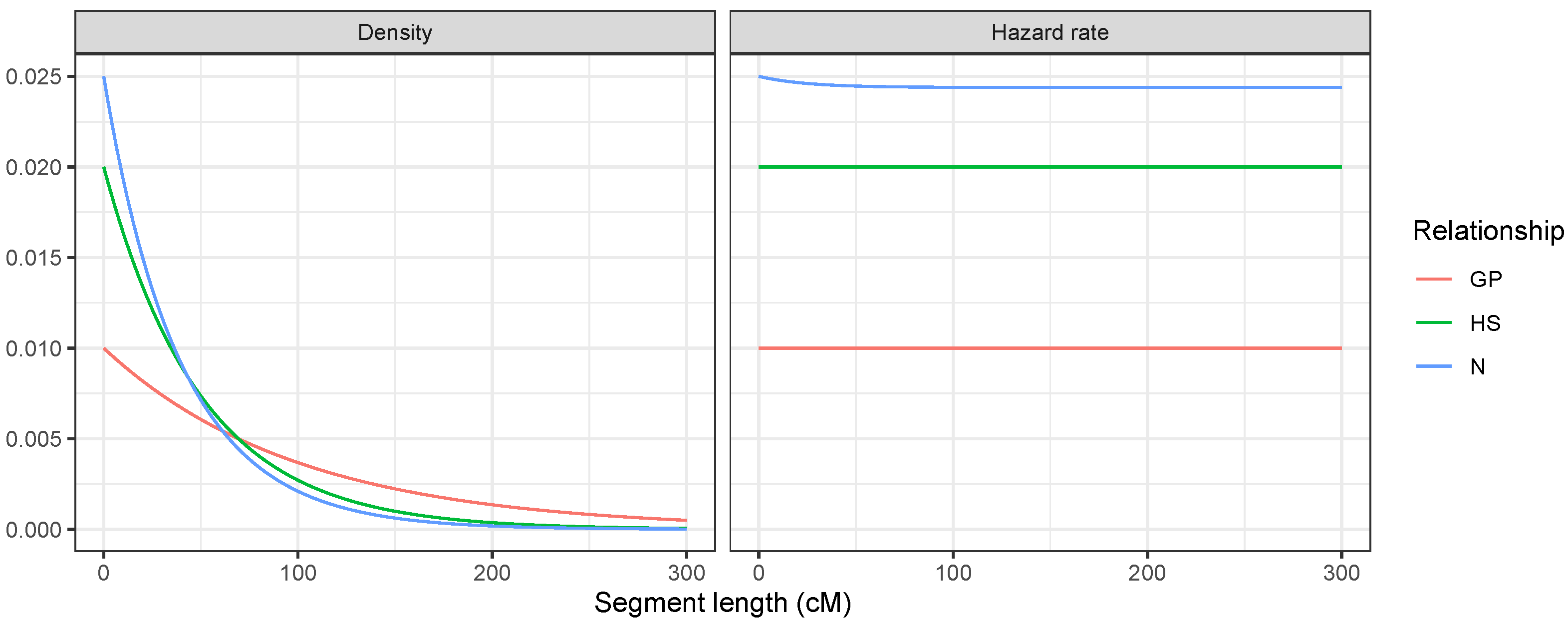
- For HS, IBD occurred if and only if the shared parent passed down the same DNA to both offspring. Two meioses could break the IBD, so the segment length followed an distribution with a constant hazard rate of 0.02.
- For GP, only one meiosis broke the IBD, so the segment length followed an distribution with a constant hazard rate of 0.01.
- For N, there were two cases. Either the parent and uncle (who are siblings) were double IBD or single IBD. If they were double IBD, then it did not matter whether the grandpaternal or grandmaternal segment was transmitted from the parent to the nephew (this did not break the segment); however, two meioses broke the segment, yielding a hazard rate of 0.02. In the single IBD case, there were three meioses that broke the segment, and the hazard rate was 0.03. Both cases were equally probable at the start of the segment, so the hazard rate was 0.025. However, as the segment progressed, the double IBD could become a single IBD and vice versa without breaking the segment. Over longer segment lengths, the probability of being in the double IBD state increased, which slightly reduced the hazard rate.Figure 3. Realised total IBD (cM) and segment count for 100,000 simulations of pedigree relationships. Some relationships with the same expected degree of IBD such as N, GP, and HS can be distinguished based on segment count. As relationships become more distant, segment count quickly becomes less useful to distinguish between relationships with the same expected degree of IBD sharing.Figure 3. Realised total IBD (cM) and segment count for 100,000 simulations of pedigree relationships. Some relationships with the same expected degree of IBD such as N, GP, and HS can be distinguished based on segment count. As relationships become more distant, segment count quickly becomes less useful to distinguish between relationships with the same expected degree of IBD sharing.
![Genes 16 00492 g003]() Figure 4. Segment length distributions at the start of a chromosome for three relationships with : grandparent–grandchild, half-siblings, and uncle–nephew.Figure 4. Segment length distributions at the start of a chromosome for three relationships with : grandparent–grandchild, half-siblings, and uncle–nephew.
Figure 4. Segment length distributions at the start of a chromosome for three relationships with : grandparent–grandchild, half-siblings, and uncle–nephew.Figure 4. Segment length distributions at the start of a chromosome for three relationships with : grandparent–grandchild, half-siblings, and uncle–nephew.![Genes 16 00492 g004]()
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
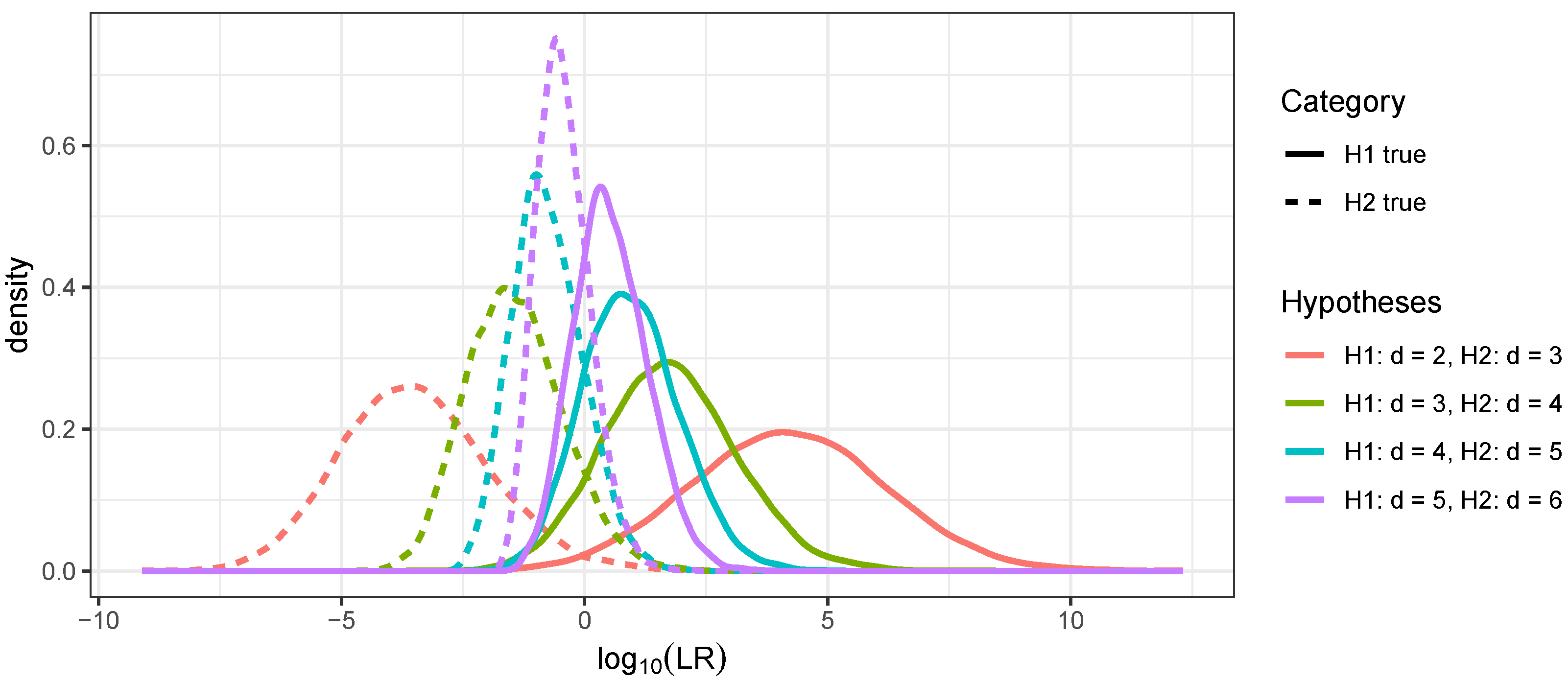
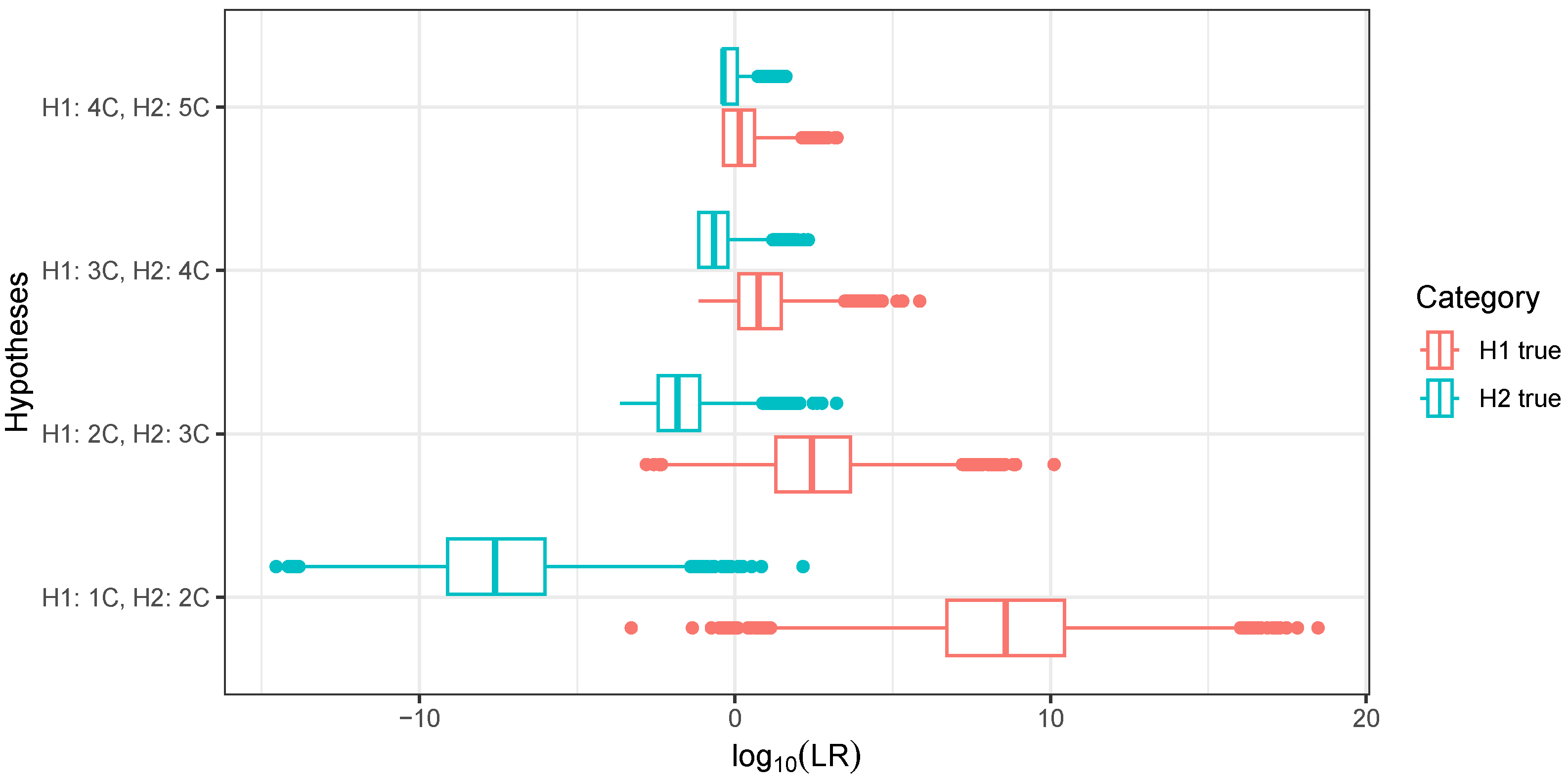
3.2. Distinguishing Linear Relationships Using Continuous IBD
3.3. Distinguishing Cousin Relationships Using Continuous IBD
3.4. Distinguishing Relationships with Identical
4. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Skare, Ø.; Sheehan, N.; Egeland, T. Identification of distant family relationships. Bioinformatics 2009, 25, 2376–2382. [Google Scholar] [CrossRef] [PubMed]
- Essen-Moller, E. Die Beweiskraft der Ahnlichkeit im Vaterschaftsnachweis-theoretische Grundlagen. Mitteilungen Anthropol. Ges. Wien 1938, 68, 9–53. [Google Scholar]
- Egeland, T.; Mostad, P.F. Statistical genetics and genetical statistics: A forensic perspective. Scand. J. Stat. 2002, 29, 297–307. [Google Scholar] [CrossRef]
- Glynn, C.L. Bridging disciplines to form a new one: The emergence of forensic genetic genealogy. Genes 2022, 13, 1381. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.F.; Murry, L.T.; Telford, L.; Moriarty, F. Direct-to-consumer genetic testing: An updated systematic review of healthcare professionals’ knowledge and views, and ethical and legal concerns. Eur. J. Hum. Genet. 2022, 30, 1331–1343. [Google Scholar] [CrossRef]
- Erlich, Y.; Shor, T.; Pe’er, I.; Carmi, S. Identity inference of genomic data using long-range familial searches. Science 2018, 362, 690–694. [Google Scholar] [CrossRef]
- Kling, D.; Phillips, C.; Kennett, D.; Tillmar, A. Investigative genetic genealogy: Current methods, knowledge and practice. Forensic Sci. Int. Genet. 2021, 52, 102474. [Google Scholar] [CrossRef]
- Weir, B.S.; Anderson, A.D.; Hepler, A.B. Genetic relatedness analysis: Modern data and new challenges. Nat. Rev. Genet. 2006, 7, 771–780. [Google Scholar] [CrossRef]
- Henn, B.M.; Hon, L.; Macpherson, J.M.; Eriksson, N.; Saxonov, S.; Pe’er, I.; Mountain, J.L. Cryptic distant relatives are common in both isolated and cosmopolitan genetic samples. PLoS ONE 2012, 7, e34267. [Google Scholar] [CrossRef]
- Tillmar, A.; Kling, D. Comparative Study of Statistical Approaches and SNP Panels to Infer Distant Relationships in Forensic Genetics. Genes 2025, 16, 114. [Google Scholar] [CrossRef]
- Balding, D.J.; Bishop, M.; Cannings, C. Handbook of Statistical Genetics; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Speed, D.; Balding, D.J. Relatedness in the post-genomic era: Is it still useful? Nat. Rev. Genet. 2015, 16, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. Inbreeding and relatedness coefficients: What do they measure? Heredity 2002, 88, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Meester, R.; Slooten, K. Probability and Forensic Evidence: Theory, Philosophy, and Applications; Cambridge University Press: Cambridge, UK, 2021. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2024. [Google Scholar]
- Lange, K. Mathematical and Statistical Methods for Genetic Analysis; Springer: Berlin/Heidelberg, Germany, 2002; Volume 488. [Google Scholar]
- Lander, E.S.; Green, P. Construction of multilocus genetic linkage maps in humans. Proc. Natl. Acad. Sci. USA 1987, 84, 2363–2367. [Google Scholar] [CrossRef]
- Kruglyak, L.; Lander, E.S. Faster multipoint linkage analysis using Fourier transforms. J. Comput. Biol. 1998, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.P. The probability that related individuals share some section of genome identical by descent. Theor. Popul. Biol. 1983, 23, 34–63. [Google Scholar] [CrossRef] [PubMed]
- Edge, M.D.; Coop, G. Donnelly (1983) and the limits of genetic genealogy. Theor. Popul. Biol. 2020, 133, 23–24. [Google Scholar] [CrossRef]
- Vigeland, M.D. Pedigree Analysis in R; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar]
- Thompson, E.A. Identity by Descent: Variation in Meiosis, Across Genomes, and in Populations. Genetics 2013, 194, 301–326. [Google Scholar] [CrossRef]
- Hill, W.G.; Weir, B.S. Variation in actual relationship as a consequence of Mendelian sampling and linkage. Genet. Res. 2011, 93, 47–64. [Google Scholar] [CrossRef]
- Balding, D.J.; Krawczak, M.; Buckleton, J.S.; Curran, J.M. Decision-making in familial database searching: KI alone or not alone? Forensic Sci. Int. Genet. 2013, 7, 52–54. [Google Scholar] [CrossRef]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef]
- Gorden, E.M.; Greytak, E.M.; Sturk-Andreaggi, K.; Cady, J.; McMahon, T.P.; Armentrout, S.; Marshall, C. Extended kinship analysis of historical remains using SNP capture. Forensic Sci. Int. Genet. 2022, 57, 102636. [Google Scholar] [CrossRef] [PubMed]
- Durand, E.Y.; Eriksson, N.; McLean, C.Y. Reducing pervasive false-positive identical-by-descent segments detected by large-scale pedigree analysis. Mol. Biol. Evol. 2014, 31, 2212–2222. [Google Scholar] [CrossRef]
- Snedecor, J.; Fennell, T.; Stadick, S.; Homer, N.; Antunes, J.; Stephens, K.; Holt, C. Fast and accurate kinship estimation using sparse SNPs in relatively large database searches. Forensic Sci. Int. Genet. 2022, 61, 102769. [Google Scholar] [CrossRef] [PubMed]
- Bettinger, B. The shared cM project: A demonstration of the power of citizen science. J. Genet. Geneal 2016, 8, 38–42. [Google Scholar]
- Gjertson, D.W.; Brenner, C.H.; Baur, M.P.; Carracedo, A.; Guidet, F.; Luque, J.A.; Lessig, R.; Mayr, W.R.; Pascali, V.L.; Prinz, M.; et al. ISFG: Recommendations on biostatistics in paternity testing. Forensic Sci. Int. Genet. 2007, 1, 223–231. [Google Scholar] [CrossRef]
- Amorim, A.; Crespillo, M.; Luque, J.A.; Prieto, L.; Garcia, O.; Gusmão, L.; Aler, M.; Barrio, P.A.; Saragoni, V.G.; Pinto, N. Formulation and communication of evaluative forensic science expert opinion—A GHEP-ISFG contribution to the establishment of standards. Forensic Sci. Int. Genet. 2016, 25, 210–213. [Google Scholar] [CrossRef]
- Kling, D.; Mostad, P.; Tillmar, A. FamLink2–A comprehensive tool for likelihood computations in pedigrees analyses involving linked DNA markers accounting for genotype uncertainties. Forensic Sci. Int. Genet. 2025, 74, 103150. [Google Scholar] [CrossRef]
- Kruijver, M. Characterizing the genetic structure of a forensic DNA database using a latent variable approach. Forensic Sci. Int. Genet. 2016, 23, 130–149. [Google Scholar] [CrossRef]
- Ramos, D.; Gonzalez-Rodriguez, J. Reliable support: Measuring calibration of likelihood ratios. Forensic Sci. Int. 2013, 230, 156–169. [Google Scholar] [CrossRef]
- Thompson, E. Gene identities and multiple relationships. Biometrics 1974, 30, 667–680. [Google Scholar] [CrossRef]
- Cox, D.R. The Theory of Stochastic Processes; Routledge: London, UK, 2017. [Google Scholar]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.; Cardon, L.R. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002, 30, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Ingolfsdottir, A.; Gudbjartsson, D. Genetic linkage analysis algorithms and their implementation. In Transactions on Computational Systems Biology III; Springer: Berlin/Heidelberg, Germany, 2005; pp. 123–144. [Google Scholar] [CrossRef]
- Halldorsson, B.V.; Palsson, G.; Stefansson, O.A.; Jonsson, H.; Hardarson, M.T.; Eggertsson, H.P.; Gunnarsson, B.; Oddsson, A.; Halldorsson, G.H.; Zink, F.; et al. Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science 2019, 363, eaau1043. [Google Scholar] [CrossRef] [PubMed]
- Vigeland, M.D. Relatedness coefficients in pedigrees with inbred founders. J. Math. Biol. 2020, 81, 185–207. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.W. Proportion of genome shared identical by descent by relatives: Concept, computation, and applications. Am. J. Hum. Genet. 1995, 56, 1468. [Google Scholar]
- Guo, S.W. Variation in genetic identity among relatives. Hum. Hered. 1996, 46, 61–70. [Google Scholar] [CrossRef]
- Vigeland, M.D. Two-locus identity coefficients in pedigrees. G3 2023, 13, jkac326. [Google Scholar] [CrossRef]
- Rabiner, L.R. A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE 1989, 77, 257–286. [Google Scholar] [CrossRef]
- Cox, D.R. Regression models and life-tables. J. R. Stat. Soc. Ser. B (Methodol.) 1972, 34, 187–202. [Google Scholar] [CrossRef]
- Klein, J.P.; Moeschberger, M.L. Survival Analysis: Techniques for Censored and Truncated Data; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
| (a) Simulated IBD Segments Only | |||
|---|---|---|---|
| Start (cM) | End (cM) | Length (cM) | IBD State () |
| 0.00 | 5.06 | 5.06 | 1 |
| 5.06 | 29.06 | 24.00 | 0 |
| 29.06 | 93.94 | 64.88 | 1 |
| 93.94 | 100.00 | 6.06 | 0 |
| (b) Unobserved IBD vector represented by an integer and the binary representation | |||
| Start (cM) | End (cM) | Length (cM) | IBD vector () |
| 0.00 | 5.06 | 5.06 | 47: [0 0 1 0 1 1 1 1] |
| 5.06 | 26.69 | 21.63 | 63: [0 0 1 1 1 1 1 1] |
| 26.69 | 27.81 | 1.12 | 191: [1 0 1 1 1 1 1 1] |
| 27.81 | 29.06 | 1.25 | 183: [1 0 1 1 0 1 1 1] |
| 29.06 | 47.96 | 18.90 | 167: [1 0 1 0 0 1 1 1] |
| 47.96 | 49.91 | 1.96 | 175: [1 0 1 0 1 1 1 1] |
| 49.91 | 78.40 | 28.49 | 167: [1 0 1 0 0 1 1 1] |
| 78.40 | 93.94 | 15.54 | 135: [1 0 0 0 0 1 1 1] |
| 93.94 | 100.00 | 6.06 | 199: [1 1 0 0 0 1 1 1] |
| Median | Accuracy | |||||
|---|---|---|---|---|---|---|
| 0.9837 | 0.0163 | 4.1709 | 0.983 | |||
| 0.0177 | 0.9823 | −3.5500 | ||||
| 0.9069 | 0.0931 | 1.7197 | 0.9123 | |||
| 0.0824 | 0.9176 | −1.4897 | ||||
| 0.8337 | 0.1663 | 0.9137 | 0.8397 | |||
| 0.1544 | 0.8456 | −0.8139 | ||||
| 0.7545 | 0.2455 | 0.4865 | 0.7743 | |||
| 0.2060 | 0.7940 | −0.4883 |
| Median | Accuracy | |||||
|---|---|---|---|---|---|---|
| 1C | 2C | 0.9986 | 0.0014 | 8.5754 | 0.9990 | |
| 0.0007 | 0.9993 | −7.6095 | ||||
| 2C | 3C | 0.9342 | 0.0658 | 2.4376 | 0.9421 | |
| 0.0501 | 0.9499 | −1.8169 | ||||
| 3C | 4C | 0.7770 | 0.2230 | 0.7456 | 0.8119 | |
| 0.1532 | 0.8468 | −0.6617 | ||||
| 4C | 5C | 0.7009 | 0.2991 | 0.1516 | 0.6980 | |
| 0.3049 | 0.6951 | −0.3655 |
| Median | Accuracy | ||||||
|---|---|---|---|---|---|---|---|
| 1/2 | GP | 0.9944 | 0.0056 | 4.7944 | 0.9946 | ||
| 0.0052 | 0.9948 | −8.1254 | |||||
| 1/2 | HS | 0.9082 | 0.0918 | 1.0630 | 0.8823 | ||
| 0.1436 | 0.8564 | −1.7842 | |||||
| 1/2 | N | 0.9023 | 0.0977 | 1.1379 | 0.8849 | ||
| 0.1324 | 0.8676 | −2.5758 | |||||
| 1/4 | 1C | 0.8761 | 0.1239 | 0.8497 | 0.8509 | ||
| 0.1742 | 0.8258 | −1.1190 | |||||
| 1/4 | GGP | 0.9503 | 0.0497 | 1.7242 | 0.9385 | ||
| 0.0734 | 0.9266 | −2.9268 | |||||
| 1/4 | GN | 0.8197 | 0.1803 | 0.3703 | 0.7465 | ||
| 0.3267 | 0.6733 | −0.3335 | |||||
| 1/4 | HN | 0.8213 | 0.1787 | 0.4690 | 0.7637 | ||
| 0.2938 | 0.7062 | −0.4700 | |||||
| 1/8 | 1C1R | 0.7938 | 0.2062 | 0.3913 | 0.7510 | ||
| 0.2918 | 0.7082 | −0.3773 | |||||
| 1/8 | GP | 0.8637 | 0.1363 | 0.7782 | 0.8390 | ||
| 0.1856 | 0.8144 | −0.9586 | |||||
| 1/8 | N | 0.7409 | 0.2591 | 0.1702 | 0.6578 | ||
| 0.4253 | 0.5747 | −0.0693 | |||||
| 1/8 | H1C | 0.7375 | 0.2625 | 0.1660 | 0.6346 | ||
| 0.4683 | 0.5317 | −0.0322 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruijver, M. An Upper Bound on the Power of DNA to Distinguish Pedigree Relationships. Genes 2025, 16, 492. https://doi.org/10.3390/genes16050492
Kruijver M. An Upper Bound on the Power of DNA to Distinguish Pedigree Relationships. Genes. 2025; 16(5):492. https://doi.org/10.3390/genes16050492
Chicago/Turabian StyleKruijver, Maarten. 2025. "An Upper Bound on the Power of DNA to Distinguish Pedigree Relationships" Genes 16, no. 5: 492. https://doi.org/10.3390/genes16050492
APA StyleKruijver, M. (2025). An Upper Bound on the Power of DNA to Distinguish Pedigree Relationships. Genes, 16(5), 492. https://doi.org/10.3390/genes16050492