
Barth Syndrome: TAFAZZIN Gene, Cardiologic Aspects, and Mitochondrial Studies—A Comprehensive Narrative Review


Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. TAFAZZIN (TAZ) Gene
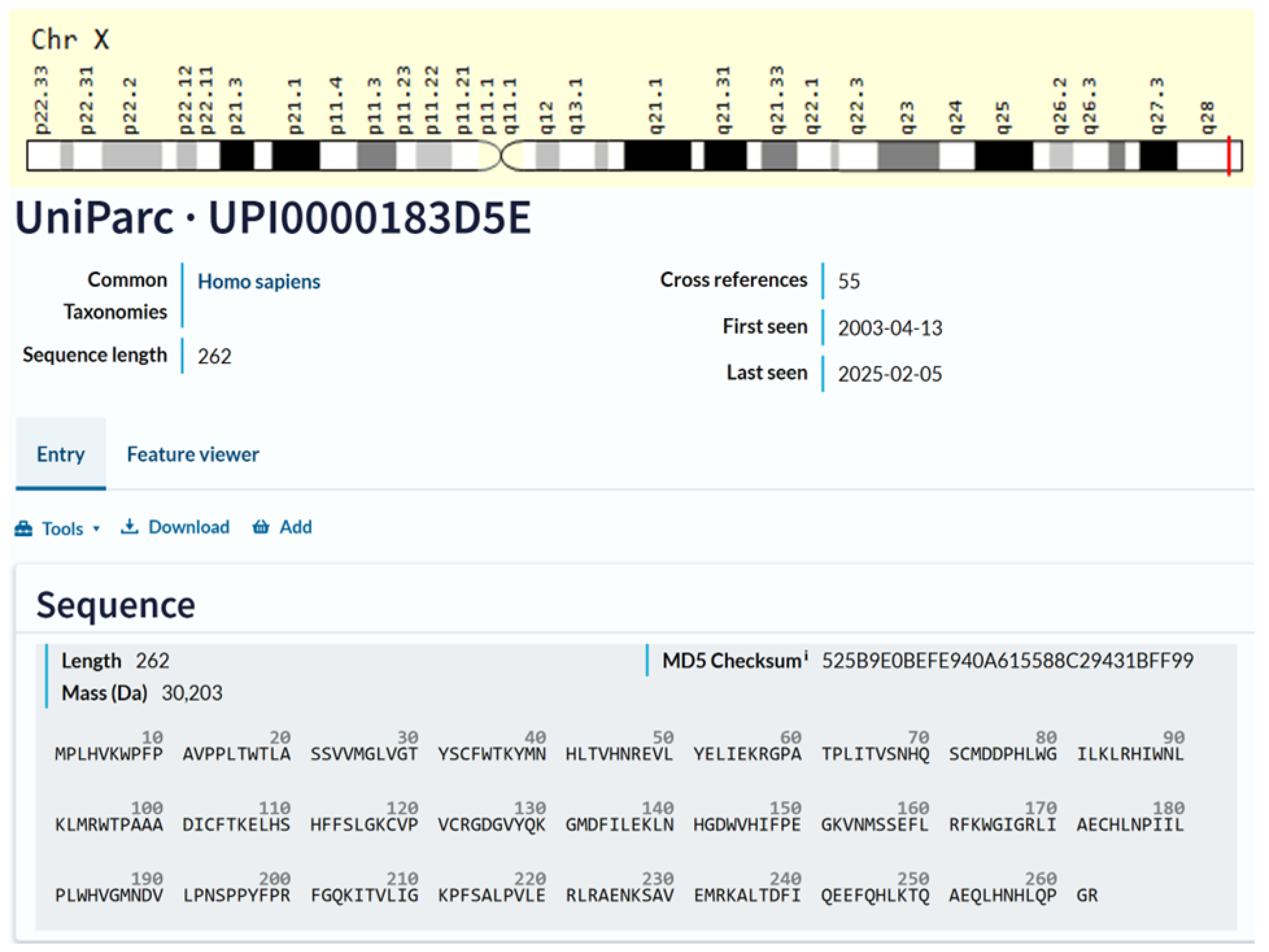
3.2. TAFAZZIN (TAZ) Protein
3.3. Animal Models
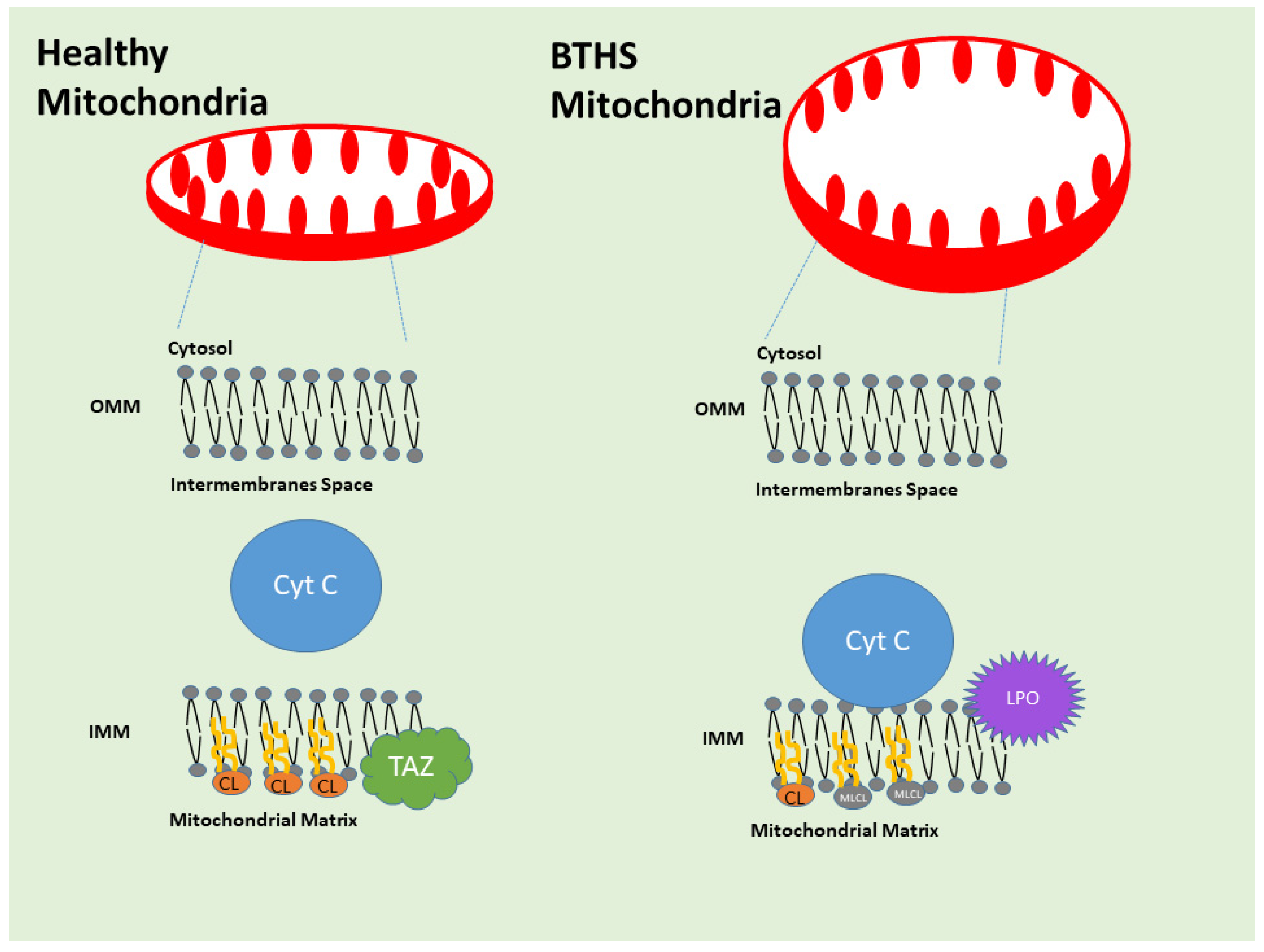
3.4. TAFAZZIN Reactome and Connectome
3.5. Barth Syndrome and Anatomic Pathology
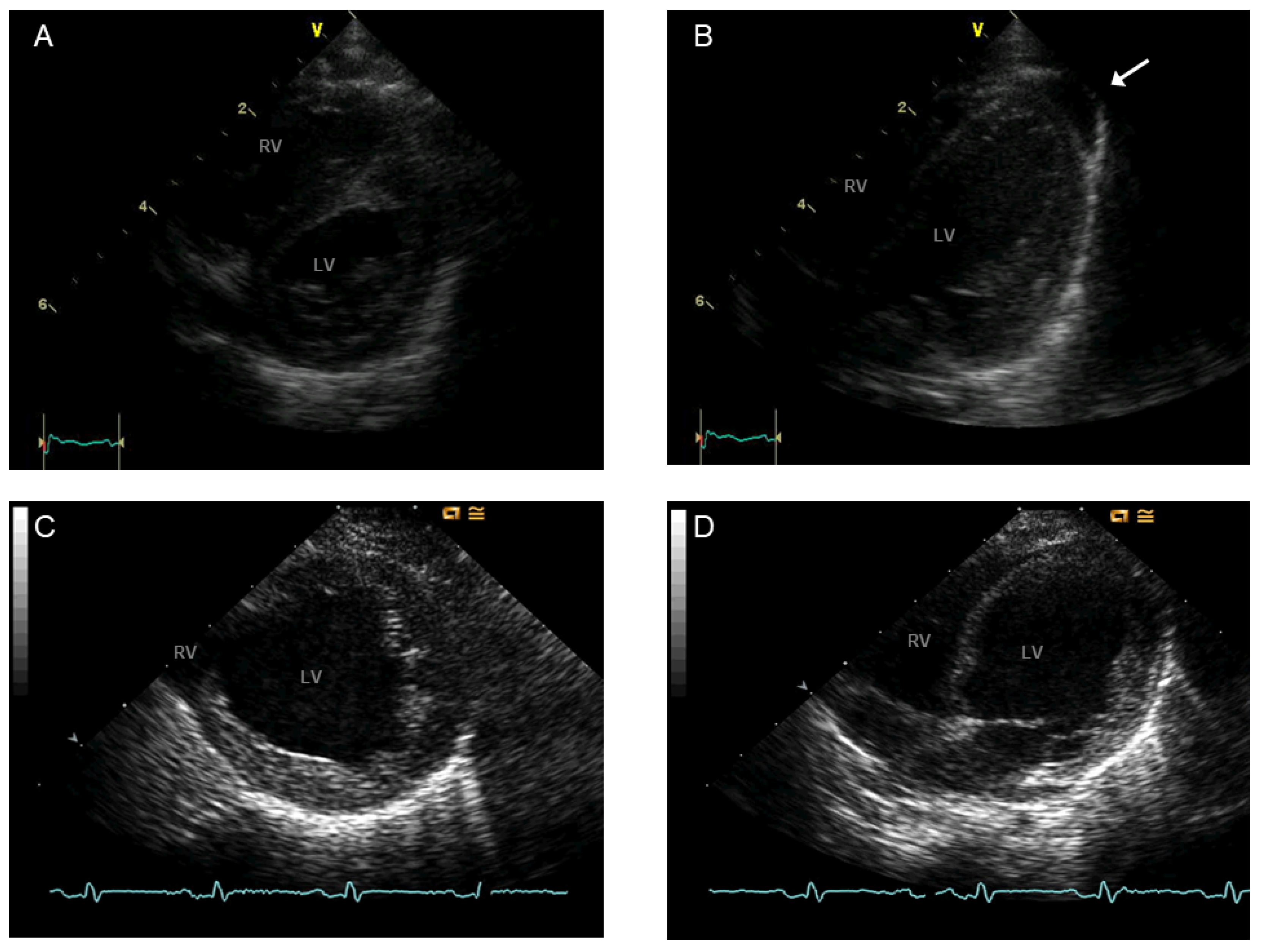
3.6. BTHS Cardiomyopathy
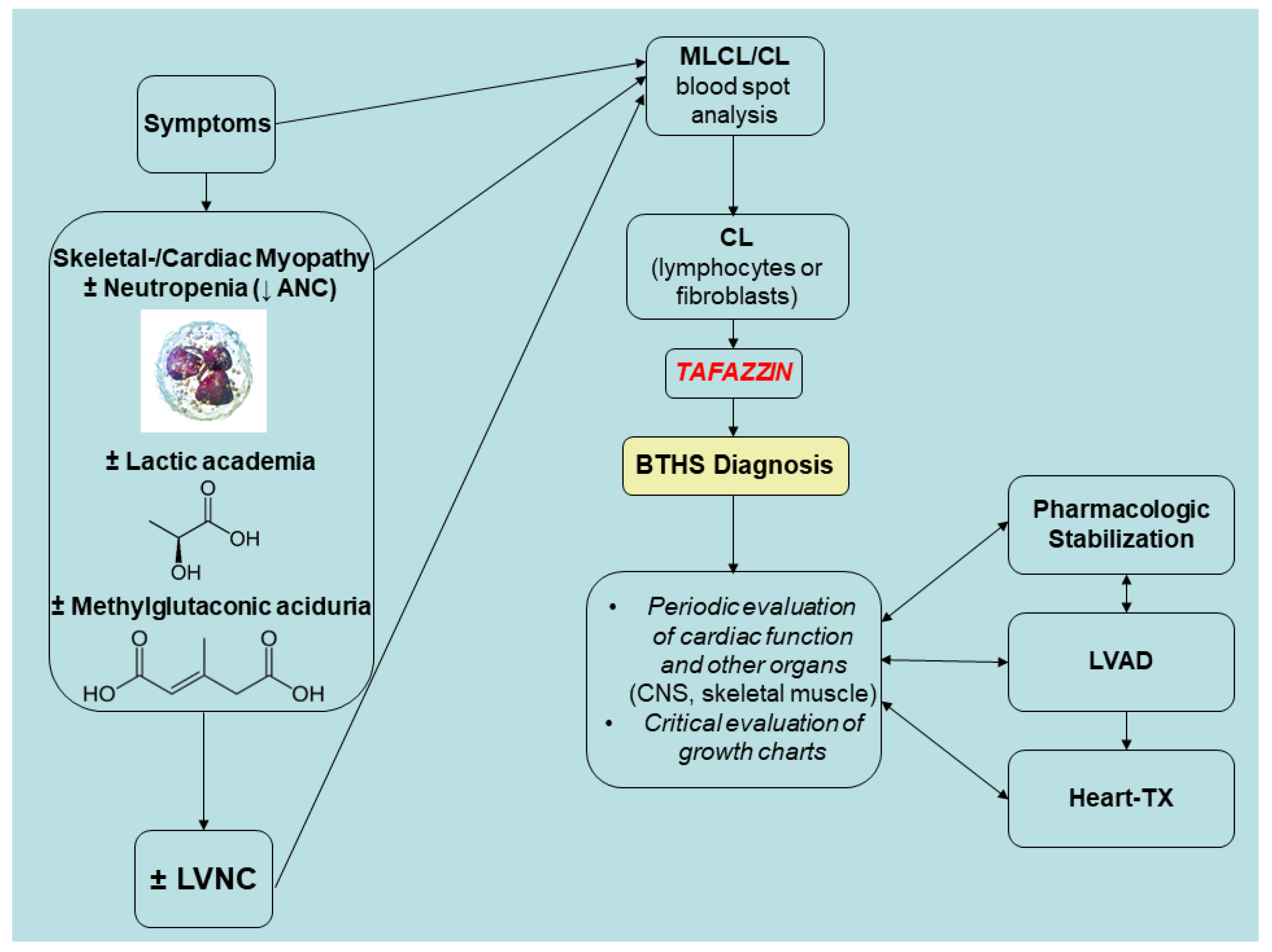
3.7. BTHS Epidemiology, Differential Diagnosis, and Prognosis
3.8. Future Directions
3.9. Conclusive Remarks
Funding
Conflicts of Interest
References
- Pang, J.; Bao, Y.; Mitchell-Silbaugh, K.; Veevers, J.; Fang, X. Barth Syndrome Cardiomyopathy: An Update. Genes 2022, 13, 656. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.; Rao, E.S.; Pierre, G.; Chronopoulou, E.; Hornby, B.; Heyman, A.; Vernon, H.J. Clinical presentation and natural history of Barth Syndrome: An overview. J. Inherit. Metab. Dis. 2022, 45, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Jefferies, J.; Wang, S.; Pu, W.T.; Takemoto, C.; Hornby, B.; Heyman, A.; Chin, M.T.; Vernon, H.J. Current and future treatment approaches for Barth syndrome. J. Inherit. Metab. Dis. 2022, 45, 17–28. [Google Scholar] [CrossRef]
- Zakrzewski, P.; Rice, C.M.; Fleming, K.; Cela, D.; Groves, S.J.; Ponce-Garcia, F.M.; Gibbs, W.; Roberts, K.; Pike, T.; Strathdee, D.; et al. Tafazzin regulates neutrophil maturation and inflammatory response. EMBO Rep. 2025, 26, 1590–1619. [Google Scholar] [CrossRef]
- Dalal, N.; Naranje, K.; Moriangthem, A.; Singh, A. Barth syndrome: A rare cause of cardiomyopathy in neonates. BMJ Case Rep. 2024, 17, e260799. [Google Scholar] [CrossRef]
- Muir, C.R.; Gilmore, K.L.; Singh, S.; Vora, N.L. Cranial, Renal, and Skeletal Anomalies in a Fetus With a Pathogenic Variant in the TAFAZZIN Gene. Prenat. Diagn. 2025, 45, 227–230. [Google Scholar] [CrossRef]
- Sniezek Carney, O.; Harris, K.W.; Wohlfarter, Y.; Lee, K.; Butschek, G.; Anzmann, A.F.; Hamacher-Brady, A.; Keller, M.A.; Vernon, H.J. Stem cell models of TAFAZZIN deficiency reveal novel tissue-specific pathologies in Barth syndrome. Hum. Mol. Genet. 2025, 34, 101–115. [Google Scholar] [CrossRef]
- Balderas, E.; Lee, S.H.J.; Rai, N.K.; Mollinedo, D.M.; Duron, H.E.; Chaudhuri, D. Mitochondrial Calcium Regulation of Cardiac Metabolism in Health and Disease. Physiology 2024, 39, 247–268. [Google Scholar] [CrossRef]
- Ferreira, C.; Pierre, G.; Thompson, R.; Vernon, H. Barth Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25299040 (accessed on 25 March 2025).
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef]
- Ades, L.C.; Gedeon, A.K.; Wilson, M.J.; Latham, M.; Partington, M.W.; Mulley, J.C.; Nelson, J.; Lui, K.; Sillence, D.O. Barth syndrome: Clinical features and confirmation of gene localisation to distal Xq28. Am. J. Med. Genet. 1993, 45, 327–334. [Google Scholar] [CrossRef]
- Bolhuis, P.A.; Hensels, G.W.; Hulsebos, T.J.; Baas, F.; Barth, P.G. Mapping of the locus for X-linked cardioskeletal myopathy with neutropenia and abnormal mitochondria (Barth syndrome) to Xq28. Am. J. Hum. Genet. 1991, 48, 481–485. [Google Scholar] [PubMed]
- Liang, Z.; Schmidtke, M.W.; Greenberg, M.L. Current Knowledge on the Role of Cardiolipin Remodeling in the Context of Lipid Oxidation and Barth Syndrome. Front. Mol. Biosci. 2022, 9, 915301. [Google Scholar] [CrossRef] [PubMed]
- Fatica, E.M.; DeLeonibus, G.A.; House, A.; Kodger, J.V.; Pearce, R.W.; Shah, R.R.; Levi, L.; Sandlers, Y. Barth Syndrome: Exploring Cardiac Metabolism with Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Metabolites 2019, 9, 306. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, P.; Fassone, L.; Gedeon, A.; Janssen, E.A.; Bione, S.; Bolhuis, P.A.; Barth, P.G.; Wilson, M.; Haan, E.; Orstavik, K.H.; et al. The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am. J. Hum. Genet. 1997, 61, 862–867. [Google Scholar] [CrossRef]
- Bleyl, S.B.; Mumford, B.R.; Brown-Harrison, M.C.; Pagotto, L.T.; Carey, J.C.; Pysher, T.J.; Ward, K.; Chin, T.K. Xq28-linked noncompaction of the left ventricular myocardium: Prenatal diagnosis and pathologic analysis of affected individuals. Am. J. Med. Genet. 1997, 72, 257–265. [Google Scholar] [CrossRef]
- Bleyl, S.B.; Mumford, B.R.; Thompson, V.; Carey, J.C.; Pysher, T.J.; Chin, T.K.; Ward, K. Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am. J. Hum. Genet. 1997, 61, 868–872. [Google Scholar] [CrossRef]
- Roberts, A.E.; Nixon, C.; Steward, C.G.; Gauvreau, K.; Maisenbacher, M.; Fletcher, M.; Geva, J.; Byrne, B.J.; Spencer, C.T. The Barth Syndrome Registry: Distinguishing disease characteristics and growth data from a longitudinal study. Am. J. Med. Genet. A 2012, 158A, 2726–2732. [Google Scholar] [CrossRef]
- Johnston, J.; Kelley, R.I.; Feigenbaum, A.; Cox, G.F.; Iyer, G.S.; Funanage, V.L.; Proujansky, R. Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am. J. Hum. Genet. 1997, 61, 1053–1058. [Google Scholar] [CrossRef]
- Cantlay, A.M.; Shokrollahi, K.; Allen, J.T.; Lunt, P.W.; Newbury-Ecob, R.A.; Steward, C.G. Genetic analysis of the G4.5 gene in families with suspected Barth syndrome. J. Pediatr. 1999, 135, 311–315. [Google Scholar] [CrossRef]
- Chen, R.; Tsuji, T.; Ichida, F.; Bowles, K.R.; Yu, X.; Watanabe, S.; Hirono, K.; Tsubata, S.; Hamamichi, Y.; Ohta, J.; et al. Mutation analysis of the G4.5 gene in patients with isolated left ventricular noncompaction. Mol. Genet. Metab. 2002, 77, 319–325. [Google Scholar] [CrossRef]
- Karall, D.; Scholl-Bürgi, S.; Sergi, C.; Geiger, R.; Engl, G.; Karall, T.; Stein, J.-I.; Vaz, F.; Schweigmann, U. Barth Syndrome and Left-Ventricular Non-Compaction: Case Report and Surveillance Plan Prior to Cardiac Transplantation. Enliven Surg. Transplant. 2014, 1, 004. [Google Scholar] [CrossRef]
- Claypool, S.M.; McCaffery, J.M.; Koehler, C.M. Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J. Cell Biol. 2006, 174, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Petit, P.X.; Ardilla-Osorio, H.; Penalvia, L.; Rainey, N.E. Tafazzin Mutation Affecting Cardiolipin Leads to Increased Mitochondrial Superoxide Anions and Mitophagy Inhibition in Barth Syndrome. Cells 2020, 9, 2333. [Google Scholar] [CrossRef]
- Schlame, M.; Xu, Y. The Function of Tafazzin, a Mitochondrial Phospholipid-Lysophospholipid Acyltransferase. J. Mol. Biol. 2020, 432, 5043–5051. [Google Scholar] [CrossRef]
- Hachmann, M.; Gulcan, G.; Rajendran, R.; Horing, M.; Liebisch, G.; Bachhuka, A.; Kohlhaas, M.; Maack, C.; Ergun, S.; Dudek, J.; et al. Tafazzin deficiency causes substantial remodeling in the lipidome of a mouse model of Barth Syndrome cardiomyopathy. Front. Mol. Med. 2024, 4, 1389456. [Google Scholar] [CrossRef]
- Oyarbide, U.; Crane, G.M.; Corey, S.J. The metabolic basis of inherited neutropenias. Br. J. Haematol. 2024, 204, 45–55. [Google Scholar] [CrossRef]
- Ji, J.; Greenberg, M.L. Cardiolipin function in the yeast S. cerevisiae and the lessons learned for Barth syndrome. J. Inherit. Metab. Dis. 2022, 45, 60–71. [Google Scholar] [CrossRef]
- Garlid, A.O.; Schaffer, C.T.; Kim, J.; Bhatt, H.; Guevara-Gonzalez, V.; Ping, P. TAZ encodes tafazzin, a transacylase essential for cardiolipin formation and central to the etiology of Barth syndrome. Gene 2020, 726, 144148. [Google Scholar] [CrossRef]
- Xu, Y.; Condell, M.; Plesken, H.; Edelman-Novemsky, I.; Ma, J.; Ren, M.; Schlame, M. A Drosophila model of Barth syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 11584–11588. [Google Scholar] [CrossRef]
- Xu, Y.; Malhotra, A.; Ren, M.; Schlame, M. The enzymatic function of tafazzin. J. Biol. Chem. 2006, 281, 39217–39224. [Google Scholar] [CrossRef]
- Acehan, D.; Malhotra, A.; Xu, Y.; Ren, M.; Stokes, D.L.; Schlame, M. Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys. J. 2011, 100, 2184–2192. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Vaz, F.; Houtkooper, R.H.; James, J.; Moore, V.; Tokunaga, C.; Kulik, W.; Wansapura, J.; Toth, M.J.; Strauss, A.; et al. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J. Biol. Chem. 2011, 286, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Soustek, M.S.; Falk, D.J.; Mah, C.S.; Toth, M.J.; Schlame, M.; Lewin, A.S.; Byrne, B.J. Characterization of a transgenic short hairpin RNA-induced murine model of Tafazzin deficiency. Hum. Gene Ther. 2011, 22, 865–871. [Google Scholar] [CrossRef]
- Wang, S.; Yazawa, E.; Keating, E.M.; Mazumdar, N.; Hauschild, A.; Ma, Q.; Wu, H.; Xu, Y.; Shi, X.; Strathdee, D.; et al. Genetic modifiers modulate phenotypic expression of tafazzin deficiency in a mouse model of Barth syndrome. Hum. Mol. Genet. 2023, 32, 2055–2067. [Google Scholar] [CrossRef]
- Xu, Y.; Kelley, R.I.; Blanck, T.J.; Schlame, M. Remodeling of cardiolipin by phospholipid transacylation. J. Biol. Chem. 2003, 278, 51380–51385. [Google Scholar] [CrossRef]
- Malhotra, A.; Edelman-Novemsky, I.; Xu, Y.; Plesken, H.; Ma, J.; Schlame, M.; Ren, M. Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proc. Natl. Acad. Sci. USA 2009, 106, 2337–2341. [Google Scholar] [CrossRef]
- Malhotra, A.; Xu, Y.; Ren, M.; Schlame, M. Formation of molecular species of mitochondrial cardiolipin. 1. A novel transacylation mechanism to shuttle fatty acids between sn-1 and sn-2 positions of multiple phospholipid species. Biochim. Biophys. Acta 2009, 1791, 314–320. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, S.; Malhotra, A.; Edelman-Novemsky, I.; Ma, J.; Kruppa, A.; Cernicica, C.; Blais, S.; Neubert, T.A.; Ren, M.; et al. Characterization of tafazzin splice variants from humans and fruit flies. J. Biol. Chem. 2009, 284, 29230–29239. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurina, Y.Y.; Mikulska-Ruminska, K.; Damschroder, D.; Vieira Neto, E.; Lasorsa, A.; Kapralov, A.A.; Tyurin, V.A.; Amoscato, A.A.; Samovich, S.N.; et al. Anomalous peroxidase activity of cytochrome c is the primary pathogenic target in Barth syndrome. Nat. Metab. 2023, 5, 2184–2205. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Bozelli, J.C., Jr.; Epand, R.M. Interplay between cardiolipin and plasmalogens in Barth syndrome. J. Inherit. Metab. Dis. 2022, 45, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Maack, C. Mechano-energetic aspects of Barth syndrome. J. Inherit. Metab. Dis. 2022, 45, 82–98. [Google Scholar] [CrossRef] [PubMed]
- Rigaud, C.; Lebre, A.S.; Touraine, R.; Beaupain, B.; Ottolenghi, C.; Chabli, A.; Ansquer, H.; Ozsahin, H.; Di Filippo, S.; De Lonlay, P.; et al. Natural history of Barth syndrome: A national cohort study of 22 patients. Orphanet J. Rare Dis. 2013, 8, 70. [Google Scholar] [CrossRef]
- Wang, J.; Guo, Y.; Huang, M.; Zhang, Z.; Zhu, J.; Liu, T.; Shi, L.; Li, F.; Huang, H.; Fu, L. Identification of TAZ mutations in pediatric patients with cardiomyopathy by targeted next-generation sequencing in a Chinese cohort. Orphanet J. Rare Dis. 2017, 12, 26. [Google Scholar] [CrossRef]
- Rodriguez, M.A.; Fernandez, L.A.; Daisley, B.A.; Reynaldi, F.J.; Allen-Vercoe, E.; Thompson, G.J. Probiotics and in-hive fermentation as a source of beneficial microbes to support the gut microbial health of honey bees. J. Insect Sci. 2023, 23, 19. [Google Scholar] [CrossRef]
- Gonzalez-Orozco, B.D.; Garcia-Cano, I.; Escobar-Zepeda, A.; Jimenez-Flores, R.; Alvarez, V.B. Metagenomic analysis and antibacterial activity of kefir microorganisms. J. Food Sci. 2023, 88, 2933–2949. [Google Scholar] [CrossRef]
- Serra, N.; Di Carlo, P.; D’Arpa, F.; Battaglia, E.; Fasciana, T.; Gulotta, G.; Maida, C.M.; Rodolico, V.; Giammanco, A.; Sergi, C. Human bile microbiota: A retrospective study focusing on age and gender. J. Infect. Public Health 2021, 14, 206–213. [Google Scholar] [CrossRef]
- Steward, C.G.; Groves, S.J.; Taylor, C.T.; Maisenbacher, M.K.; Versluys, B.; Newbury-Ecob, R.A.; Ozsahin, H.; Damin, M.K.; Bowen, V.M.; McCurdy, K.R.; et al. Neutropenia in Barth syndrome: Characteristics, risks, and management. Curr. Opin. Hematol. 2019, 26, 6–15. [Google Scholar] [CrossRef]
- Villalba-Orero, M.; Lopez-Olaneta, M.M.; Gonzalez-Lopez, E.; Padron-Barthe, L.; Gomez-Salinero, J.M.; Garcia-Prieto, J.; Wai, T.; Garcia-Pavia, P.; Ibanez, B.; Jimenez-Borreguero, L.J.; et al. Lung ultrasound as a translational approach for non-invasive assessment of heart failure with reduced or preserved ejection fraction in mice. Cardiovasc. Res. 2017, 113, 1113–1123. [Google Scholar] [CrossRef]
- Kang, S.L.; Forsey, J.; Dudley, D.; Steward, C.G.; Tsai-Goodman, B. Clinical Characteristics and Outcomes of Cardiomyopathy in Barth Syndrome: The UK Experience. Pediatr. Cardiol. 2016, 37, 167–176. [Google Scholar] [CrossRef]
- Thompson, W.R.; DeCroes, B.; McClellan, R.; Rubens, J.; Vaz, F.M.; Kristaponis, K.; Avramopoulos, D.; Vernon, H.J. New targets for monitoring and therapy in Barth syndrome. Genet. Med. 2016, 18, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.G.; Clayton, P.E. Disorders of Growth Hormone in Childhood. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25905205 (accessed on 25 March 2025).
- Greenwell, A.A.; Tabatabaei Dakhili, S.A.; Ussher, J.R. Myocardial disturbances of intermediary metabolism in Barth syndrome. Front. Cardiovasc. Med. 2022, 9, 981972. [Google Scholar] [CrossRef] [PubMed]
- Ikon, N.; Ryan, R.O. Barth Syndrome: Connecting Cardiolipin to Cardiomyopathy. Lipids 2017, 52, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Greenwell, A.A.; Gopal, K.; Altamimi, T.R.; Saed, C.T.; Wang, F.; Tabatabaei Dakhili, S.A.; Ho, K.L.; Zhang, L.; Eaton, F.; Kruger, J.; et al. Barth syndrome-related cardiomyopathy is associated with a reduction in myocardial glucose oxidation. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2255–H2269. [Google Scholar] [CrossRef]
- Thiels, C.; Fleger, M.; Huemer, M.; Rodenburg, R.J.; Vaz, F.M.; Houtkooper, R.H.; Haack, T.B.; Prokisch, H.; Feichtinger, R.G.; Lücke, T.; et al. Atypical Clinical Presentations of TAZ Mutations: An Underdiagnosed Cause of Growth Retardation? JIMD Rep. 2016, 29, 89–93. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Clarke, S.L.; Bowron, A.; Gonzalez, I.L.; Groves, S.J.; Newbury-Ecob, R.; Clayton, N.; Martin, R.P.; Tsai-Goodman, B.; Garratt, V.; Ashworth, M.; et al. Barth syndrome. Orphanet J. Rare Dis. 2013, 8, 23. [Google Scholar] [CrossRef]
- Takeda, A.; Sudo, A.; Yamada, M.; Yamazawa, H.; Izumi, G.; Nishino, I.; Ariga, T. Eponym: Barth syndrome. Eur. J. Pediatr. 2011, 170, 1365–1367. [Google Scholar] [CrossRef]
- Takeda, A.; Sudo, A.; Yamada, M.; Yamazawa, H.; Izumi, G.; Nishino, I.; Ariga, T. Barth syndrome diagnosed in the subclinical stage of heart failure based on the presence of lipid storage myopathy and isolated noncompaction of the ventricular myocardium. Eur. J. Pediatr. 2011, 170, 1481–1484. [Google Scholar] [CrossRef]
- Baban, A.; Adorisio, R.; Corica, B.; Rizzo, C.; Cali, F.; Semeraro, M.; Taurisano, R.; Magliozzi, M.; Carrozzo, R.; Parisi, F.; et al. Delayed appearance of 3-methylglutaconic aciduria in neonates with early onset metabolic cardiomyopathies: A potential pitfall for the diagnosis. Am. J. Med. Genet. A 2020, 182, 64–70. [Google Scholar] [CrossRef]
- Vernon, H.J.; Sandlers, Y.; McClellan, R.; Kelley, R.I. Clinical laboratory studies in Barth Syndrome. Mol. Genet. Metab. 2014, 112, 143–147. [Google Scholar] [CrossRef]
- Miller, P.C.; Ren, M.; Schlame, M.; Toth, M.J.; Phoon, C.K.L. A Bayesian Analysis to Determine the Prevalence of Barth Syndrome in the Pediatric Population. J. Pediatr. 2020, 217, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Garfinkel, A.C.; Seidman, J.G.; Seidman, C.E. Genetic Pathogenesis of Hypertrophic and Dilated Cardiomyopathy. Heart Fail. Clin. 2018, 14, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.T.; Byrne, B.J.; Bryant, R.M.; Margossian, R.; Maisenbacher, M.; Breitenger, P.; Benni, P.B.; Redfearn, S.; Marcus, E.; Cade, W.T. Impaired cardiac reserve and severely diminished skeletal muscle O2 utilization mediate exercise intolerance in Barth syndrome. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2122–H2129. [Google Scholar] [CrossRef]
- Mangat, J.; Lunnon-Wood, T.; Rees, P.; Elliott, M.; Burch, M. Successful cardiac transplantation in Barth syndrome--single-centre experience of four patients. Pediatr. Transplant. 2007, 11, 327–331. [Google Scholar] [CrossRef]
- Schranz, D.; Krause, U.; Kerst, G.; Esmaeili, A.; Paul, T. Functional regeneration of dilated cardiomyopathy by transcatheter bilateral pulmonary artery banding: First-in-human case series. Eur. Heart J. Case Rep. 2023, 7, ytad052. [Google Scholar] [CrossRef]
- Chowdhury, S.; Jackson, L.; Byrne, B.J.; Bryant, R.M.; Cade, W.T.; Churchill, T.L.; Buchanan, J.; Taylor, C. Longitudinal Observational Study of Cardiac Outcome Risk Factor Prediction in Children, Adolescents, and Adults with Barth Syndrome. Pediatr. Cardiol. 2022, 43, 1251–1263. [Google Scholar] [CrossRef]
- Lodato, V.; Parlapiano, G.; Cali, F.; Silvetti, M.S.; Adorisio, R.; Armando, M.; El Hachem, M.; Romanzo, A.; Dionisi-Vici, C.; Digilio, M.C.; et al. Cardiomyopathies in Children and Systemic Disorders When Is It Useful to Look beyond the Heart? J. Cardiovasc. Dev. Dis. 2022, 9, 47. [Google Scholar] [CrossRef]
- Li, Y.; Godown, J.; Taylor, C.L.; Dipchand, A.I.; Bowen, V.M.; Feingold, B. Favorable outcomes after heart transplantation in Barth syndrome. J. Heart Lung Transplant. 2021, 40, 1191–1198. [Google Scholar] [CrossRef]
- Parisi, X.; Bledsoe, J.R. Discerning clinicopathological features of congenital neutropenia syndromes: An approach to diagnostically challenging differential diagnoses. J. Clin. Pathol. 2024, 77, 586–604. [Google Scholar] [CrossRef]
- Barth, P.G.; Scholte, H.R.; Berden, J.A.; Van der Klei-Van Moorsel, J.M.; Luyt-Houwen, I.E.; Van’t Veer-Korthof, E.T.; Van der Harten, J.J.; Sobotka-Plojhar, M.A. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983, 62, 327–355. [Google Scholar] [CrossRef]
- Gedeon, A.K.; Wilson, M.J.; Colley, A.C.; Sillence, D.O.; Mulley, J.C. X linked fatal infantile cardiomyopathy maps to Xq28 and is possibly allelic to Barth syndrome. J. Med. Genet. 1995, 32, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Hornby, B.; McClellan, R.; Buckley, L.; Carson, K.; Gooding, T.; Vernon, H.J. Functional exercise capacity, strength, balance and motion reaction time in Barth syndrome. Orphanet J. Rare Dis. 2019, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Steward, C.G.; Newbury-Ecob, R.A.; Hastings, R.; Smithson, S.F.; Tsai-Goodman, B.; Quarrell, O.W.; Kulik, W.; Wanders, R.; Pennock, M.; Williams, M.; et al. Barth syndrome: An X-linked cause of fetal cardiomyopathy and stillbirth. Prenat. Diagn. 2010, 30, 970–976. [Google Scholar] [CrossRef]
- Hastings, R.; Steward, C.; Tsai-Goodman, B.; Newbury-Ecob, R. Dysmorphology of Barth syndrome. Clin. Dysmorphol. 2009, 18, 185–187. [Google Scholar] [CrossRef]
- Mazzocco, M.M.; Henry, A.E.; Kelly, R.I. Barth syndrome is associated with a cognitive phenotype. J. Dev. Behav. Pediatr. 2007, 28, 22–30. [Google Scholar] [CrossRef]
- Raches, D.; Mazzocco, M.M. Emergence and nature of mathematical difficulties in young children with Barth syndrome. J. Dev. Behav. Pediatr. 2012, 33, 328–335. [Google Scholar] [CrossRef]
- Reynolds, S.; Kreider, C.M.; Bendixen, R. A mixed-methods investigation of sensory response patterns in Barth syndrome: A clinical phenotype? Am. J. Med. Genet. A 2012, 158A, 1647–1653. [Google Scholar] [CrossRef]
- Storch, E.A.; Keeley, M.; Merlo, L.J.; St Amant, J.B.; Jacob, M.; Storch, J.F.; Spencer, C.; Byrne, B.J. Psychosocial Functioning in Youth with Barth Syndrome. Child. Health Care 2009, 38, 137–156. [Google Scholar] [CrossRef]
- Donati, M.A.; Malvagia, S.; Pasquini, E.; Morrone, A.; La Marca, G.; Garavaglia, B.; Toniolo, D.; Zammarchi, E. Barth syndrome presenting with acute metabolic decompensation in the neonatal period. J. Inherit. Metab. Dis. 2006, 29, 684. [Google Scholar] [CrossRef]
- Yen, T.Y.; Hwu, W.L.; Chien, Y.H.; Wu, M.H.; Lin, M.T.; Tsao, L.Y.; Hsieh, W.S.; Lee, N.C. Acute metabolic decompensation and sudden death in Barth syndrome: Report of a family and a literature review. Eur. J. Pediatr. 2008, 167, 941–944. [Google Scholar] [CrossRef]
- Barth, R.F.; Buja, L.M.; Barth, A.L.; Carpenter, D.E.; Parwani, A.V. A Comparison of the Clinical, Viral, Pathologic, and Immunologic Features of Severe Acute Respiratory Syndrome (SARS), Middle East Respiratory Syndrome (MERS), and Coronavirus 2019 (COVID-19) Diseases. Arch. Pathol. Lab. Med. 2021, 145, 1194–1211. [Google Scholar] [CrossRef] [PubMed]
- van Werkhoven, M.A.; Thorburn, D.R.; Gedeon, A.K.; Pitt, J.J. Monolysocardiolipin in cultured fibroblasts is a sensitive and specific marker for Barth Syndrome. J. Lipid Res. 2006, 47, 2346–2351. [Google Scholar] [CrossRef] [PubMed]
- Kulik, W.; van Lenthe, H.; Stet, F.S.; Houtkooper, R.H.; Kemp, H.; Stone, J.E.; Steward, C.G.; Wanders, R.J.; Vaz, F.M. Bloodspot assay using HPLC-tandem mass spectrometry for detection of Barth syndrome. Clin. Chem. 2008, 54, 371–378. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Rodenburg, R.J.; Thiels, C.; van Lenthe, H.; Stet, F.; Poll-The, B.T.; Stone, J.E.; Steward, C.G.; Wanders, R.J.; Smeitink, J.; et al. Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography-mass spectrometry as a diagnostic test for Barth syndrome. Anal. Biochem. 2009, 387, 230–237. [Google Scholar] [CrossRef]
- Spencer, C.T.; Bryant, R.M.; Day, J.; Gonzalez, I.L.; Colan, S.D.; Thompson, W.R.; Berthy, J.; Redfearn, S.P.; Byrne, B.J. Cardiac and clinical phenotype in Barth syndrome. Pediatrics 2006, 118, e337-346. [Google Scholar] [CrossRef]
- Christodoulou, J.; McInnes, R.R.; Jay, V.; Wilson, G.; Becker, L.E.; Lehotay, D.C.; Platt, B.A.; Bridge, P.J.; Robinson, B.H.; Clarke, J.T. Barth syndrome: Clinical observations and genetic linkage studies. Am. J. Med. Genet. 1994, 50, 255–264. [Google Scholar] [CrossRef]
- Barth, P.G.; Van den Bogert, C.; Bolhuis, P.A.; Scholte, H.R.; van Gennip, A.H.; Schutgens, R.B.; Ketel, A.G. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): Respiratory-chain abnormalities in cultured fibroblasts. J. Inherit. Metab. Dis. 1996, 19, 157–160. [Google Scholar] [CrossRef]
- Wanders, R.J.; Heymans, H.S.; Schutgens, R.B.; Barth, P.G.; van den Bosch, H.; Tager, J.M. Peroxisomal disorders in neurology. J. Neurol. Sci. 1988, 88, 1–39. [Google Scholar] [CrossRef]
- Pineiro-Llanes, J.; Suzuki-Hatano, S.; Jain, A.; Perez Medina, V.A.; Cade, W.T.; Pacak, C.A.; Simmons, C.S. Matrix produced by diseased cardiac fibroblasts affects early myotube formation and function. Acta Biomater. 2022, 152, 100–112. [Google Scholar] [CrossRef]
- De Kremer, R.D.; Paschini-Capra, A.; Bacman, S.; Argarana, C.; Civallero, G.; Kelley, R.I.; Guelbert, N.; Latini, A.; Noher de Halac, I.; Giner-Ayala, A.; et al. Barth’s syndrome-like disorder: A new phenotype with a maternally inherited A3243G substitution of mitochondrial DNA (MELAS mutation). Am. J. Med. Genet. 2001, 99, 83–93. [Google Scholar] [CrossRef]
- Orstavik, K.H.; Orstavik, R.E.; Naumova, A.K.; D’Adamo, P.; Gedeon, A.; Bolhuis, P.A.; Barth, P.G.; Toniolo, D. X chromosome inactivation in carriers of Barth syndrome. Am. J. Hum. Genet. 1998, 63, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Cosson, L.; Toutain, A.; Simard, G.; Kulik, W.; Matyas, G.; Guichet, A.; Blasco, H.; Maakaroun-Vermesse, Z.; Vaillant, M.C.; Le Caignec, C.; et al. Barth syndrome in a female patient. Mol. Genet. Metab. 2012, 106, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Avdjieva-Tzavella, D.M.; Todorova, A.P.; Kathom, H.M.; Ivanova, M.B.; Yordanova, I.T.; Todorov, T.P.; Litvinenko, I.O.; Dasheva-Dimitrova, A.T.; Tincheva, R.S. Barth Syndrome in Male and Female Siblings Caused by a Novel Mutation in the Taz Gene. Genet. Couns. 2016, 27, 495–501. [Google Scholar]
- Bissler, J.J.; Tsoras, M.; Goring, H.H.; Hug, P.; Chuck, G.; Tombragel, E.; McGraw, C.; Schlotman, J.; Ralston, M.A.; Hug, G. Infantile dilated X-linked cardiomyopathy, G4.5 mutations, altered lipids, and ultrastructural malformations of mitochondria in heart, liver, and skeletal muscle. Lab. Investig. 2002, 82, 335–344. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Turkenburg, M.; Poll-The, B.T.; Karall, D.; Perez-Cerda, C.; Morrone, A.; Malvagia, S.; Wanders, R.J.; Kulik, W.; Vaz, F.M. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta 2009, 1788, 2003–2014. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Vaz, F.M. Cardiolipin, the heart of mitochondrial metabolism. Cell Mol. Life Sci. 2008, 65, 2493–2506. [Google Scholar] [CrossRef]
- Le, C.H.; Benage, L.G.; Specht, K.S.; Li Puma, L.C.; Mulligan, C.M.; Heuberger, A.L.; Prenni, J.E.; Claypool, S.M.; Chatfield, K.C.; Sparagna, G.C.; et al. Tafazzin deficiency impairs CoA-dependent oxidative metabolism in cardiac mitochondria. J. Biol. Chem. 2020, 295, 12485–12497. [Google Scholar] [CrossRef]
- Kiebish, M.A.; Yang, K.; Liu, X.; Mancuso, D.J.; Guan, S.; Zhao, Z.; Sims, H.F.; Cerqua, R.; Cade, W.T.; Han, X.; et al. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 2013, 54, 1312–1325. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Chen, Z.; Zhu, M.; Shen, Y.; Leon, L.J.; Chi, L.; Spinozzi, S.; Tan, C.; Gu, Y.; Nguyen, A.; et al. Cardiolipin Remodeling Defects Impair Mitochondrial Architecture and Function in a Murine Model of Barth Syndrome Cardiomyopathy. Circ. Heart Fail. 2021, 14, e008289. [Google Scholar] [CrossRef]
- Imai-Okazaki, A.; Kishita, Y.; Kohda, M.; Yatsuka, Y.; Hirata, T.; Mizuno, Y.; Harashima, H.; Hirono, K.; Ichida, F.; Noguchi, A.; et al. Barth Syndrome: Different Approaches to Diagnosis. J. Pediatr. 2018, 193, 256–260. [Google Scholar] [CrossRef]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Loshuertos, R.; Fernandez-Silva, P. Isolation of Mitochondria for Mitochondrial Supercomplex Analysis from Small Tissue and Cell Culture Samples. J. Vis. Exp. 2024, 207, e66771. [Google Scholar] [CrossRef] [PubMed]
- Corey, R.A.; Harrison, N.; Stansfeld, P.J.; Sansom, M.S.P.; Duncan, A.L. Cardiolipin, and not monolysocardiolipin, preferentially binds to the interface of complexes III and IV. Chem. Sci. 2022, 13, 13489–13498. [Google Scholar] [CrossRef]
- Wang, E.; Specht, K.S.; Chicco, A.J.; Wilson, J.W. High-Repetition-Rate Transient Absorption Spectroscopy of Respiratory Supercomplexes. J. Phys. Chem. B 2022, 126, 1404–1412. [Google Scholar] [CrossRef]
- Raja, V.; Salsaa, M.; Joshi, A.S.; Li, Y.; van Roermund, C.W.T.; Saadat, N.; Lazcano, P.; Schmidtke, M.; Huttemann, M.; Gupta, S.V.; et al. Cardiolipin-deficient cells depend on anaplerotic pathways to ameliorate defective TCA cycle function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 654–661. [Google Scholar] [CrossRef]
- Raja, V.; Joshi, A.S.; Li, G.; Maddipati, K.R.; Greenberg, M.L. Loss of Cardiolipin Leads to Perturbation of Acetyl-CoA Synthesis. J. Biol. Chem. 2017, 292, 1092–1102. [Google Scholar] [CrossRef]
- Pineiro-Llanes, J.; Suzuki-Hatano, S.; Jain, A.; Venigalla, S.; Kamat, M.; Basso, K.B.; Cade, W.T.; Simmons, C.S.; Pacak, C.A. Rescue of mitochondrial dysfunction through alteration of extracellular matrix composition in barth syndrome cardiac fibroblasts. Biomaterials 2025, 315, 122922. [Google Scholar] [CrossRef]
- Liang, Z.; Ralph-Epps, T.; Schmidtke, M.W.; Lazcano, P.; Denis, S.W.; Balazova, M.; Teixeira da Rosa, N., Jr.; Chakkour, M.; Hazime, S.; Ren, M.; et al. Upregulation of the AMPK-FOXO1-PDK4 pathway is a primary mechanism of pyruvate dehydrogenase activity reduction in tafazzin-deficient cells. Sci. Rep. 2024, 14, 11497. [Google Scholar] [CrossRef]
- Snider, P.L.; Sierra Potchanant, E.A.; Sun, Z.; Edwards, D.M.; Chan, K.K.; Matias, C.; Awata, J.; Sheth, A.; Pride, P.M.; Payne, R.M.; et al. A Barth Syndrome Patient-Derived D75H Point Mutation in TAFAZZIN Drives Progressive Cardiomyopathy in Mice. Int. J. Mol. Sci. 2024, 25, 8201. [Google Scholar] [CrossRef]
- Brault, J.J.; Conway, S.J. What can ATP content tell us about Barth syndrome muscle phenotypes? J. Transl. Genet. Genom. 2025, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bertero, E.; Nickel, A.; Kohlhaas, M.; Hohl, M.; Sequeira, V.; Brune, C.; Schwemmlein, J.; Abesser, M.; Schuh, K.; Kutschka, I.; et al. Loss of Mitochondrial Ca2+ Uniporter Limits Inotropic Reserve and Provides Trigger and Substrate for Arrhythmias in Barth Syndrome Cardiomyopathy. Circulation 2021, 144, 1694–1713. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Zulkifli, M.; Joshi, A.; Venkatesan, M.; Cristel, A.; Vishnu, N.; Madesh, M.; Gohil, V.M. MCU-complex-mediated mitochondrial calcium signaling is impaired in Barth syndrome. Hum. Mol. Genet. 2022, 31, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Basu Ball, W.; Madaris, T.R.; Srikantan, S.; Madesh, M.; Mootha, V.K.; Gohil, V.M. An essential role for cardiolipin in the stability and function of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. USA 2020, 117, 16383–16390. [Google Scholar] [CrossRef]
- Jalmar, O.; Francois-Moutal, L.; Garcia-Saez, A.J.; Perry, M.; Granjon, T.; Gonzalvez, F.; Gottlieb, E.; Ayala-Sanmartin, J.; Klosgen, B.; Schwille, P.; et al. Caspase-8 binding to cardiolipin in giant unilamellar vesicles provides a functional docking platform for bid. PLoS ONE 2013, 8, e55250. [Google Scholar] [CrossRef]
- Monteiro, J.P.; Oliveira, P.J.; Jurado, A.S. Mitochondrial membrane lipid remodeling in pathophysiology: A new target for diet and therapeutic interventions. Prog. Lipid Res. 2013, 52, 513–528. [Google Scholar] [CrossRef]
- Sohn, J.; Milosevic, J.; Brouse, T.; Aziz, N.; Elkhoury, J.; Wang, S.; Hauschild, A.; van Gastel, N.; Cetinbas, M.; Tufa, S.F.; et al. A new murine model of Barth syndrome neutropenia links TAFAZZIN deficiency to increased ER stress-induced apoptosis. Blood Adv. 2022, 6, 2557–2577. [Google Scholar] [CrossRef]
- Colina-Tenorio, L.; Horten, P.; Pfanner, N.; Rampelt, H. Shaping the mitochondrial inner membrane in health and disease. J. Intern. Med. 2020, 287, 645–664. [Google Scholar] [CrossRef]
- Duncan, A.L. Monolysocardiolipin (MLCL) interactions with mitochondrial membrane proteins. Biochem. Soc. Trans. 2020, 48, 993–1004. [Google Scholar] [CrossRef]
- Ruiz-Ramirez, A.; Barrios-Maya, M.; Quezada-Pablo, H.; Lopez-Acosta, O.; El-Hafidi, M. Kidney dysfunction induced by a sucrose-rich diet in rat involves mitochondria ROS generation, cardiolipin changes, and the decline of autophagy protein markers. Am. J. Physiol. Renal Physiol. 2020, 318, F53–F66. [Google Scholar] [CrossRef]
- Dudek, J.; Hartmann, M.; Rehling, P. The role of mitochondrial cardiolipin in heart function and its implication in cardiac disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 810–821. [Google Scholar] [CrossRef]
- Dinca, A.A.; Chien, W.M.; Chin, M.T. Identification of novel mitochondrial localization signals in human Tafazzin, the cause of the inherited cardiomyopathic disorder Barth syndrome. J. Mol. Cell Cardiol. 2018, 114, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, K.; Fujioka, H.; Tandler, B.; Hoppel, C.L. Cardiac mitochondrial structure and function in tafazzin-knockdown mice. Mitochondrion 2018, 43, 53–62. [Google Scholar] [CrossRef]
- Ikon, N.; Su, B.; Hsu, F.F.; Forte, T.M.; Ryan, R.O. Exogenous cardiolipin localizes to mitochondria and prevents TAZ knockdown-induced apoptosis in myeloid progenitor cells. Biochem. Biophys. Res. Commun. 2015, 464, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Stollberger, C. Ultrastructural findings in noncompaction prevail with neuromuscular disorders. Cardiology 2013, 126, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Sniezek Carney, O.; Harris, K.W.; Wohlfarter, Y.; Lee, K.; Butschek, G.; Anzmann, A.; Claypool, S.M.; Hamacher-Brady, A.; Keller, M.; Vernon, H.J. Stem cell models of TAFAZZIN deficiency reveal novel tissue-specific pathologies in Barth Syndrome. bioRxiv 2024. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Nie, J.; Shi, Y. Restoration of mitophagy ameliorates cardiomyopathy in Barth syndrome. Autophagy 2022, 18, 2134–2149. [Google Scholar] [CrossRef]
- Jantuan, E.; Chiu, B.; Chiu, B.; Shen, F.; Oudit, G.Y.; Sergi, C. The tumor microenvironment may trigger lymphoproliferation in cardiac myxoma. Transl. Oncol. 2021, 14, 100911. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Espeel, M.; Almeida, L.; Reimer, A.; Bosboom, D.; Roels, F.; de Brouwer, A.P.; Wevers, R.A. Inborn errors of metabolism in the biosynthesis and remodelling of phospholipids. J. Inherit. Metab. Dis. 2015, 38, 99–110. [Google Scholar] [CrossRef]
- Tovaglieri, N.; Russo, S.; Micaglio, E.; Corcelli, A.; Lobasso, S. Case report: Variability in clinical features as a potential pitfall for the diagnosis of Barth syndrome. Front. Pediatr. 2023, 11, 1250772. [Google Scholar] [CrossRef]
- Beecher, G.; Fleming, M.D.; Liewluck, T. Hereditary myopathies associated with hematological abnormalities. Muscle Nerve 2022, 65, 374–390. [Google Scholar] [CrossRef]
- Mauritz, M.D.; Hasan, C.; Schreiber, L.; Wegener-Panzer, A.; Barth, S.; Zernikow, B. Differential Diagnosis of Cyclic Vomiting and Periodic Headaches in a Child with Ventriculoperitoneal Shunt: Case Report of Chronic Shunt Overdrainage. Children 2022, 9, 432. [Google Scholar] [CrossRef] [PubMed]
- Lodi, L.; Melki, I.; Bondet, V.; Seabra, L.; Rice, G.I.; Carter, E.; Lepelley, A.; Martin-Niclos, M.J.; Al Adba, B.; Bader-Meunier, B.; et al. Differential Expression of Interferon-Alpha Protein Provides Clues to Tissue Specificity Across Type I Interferonopathies. J. Clin. Immunol. 2021, 41, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.; Wagner, S.; Hammond, J.; Roberts, N.; Marshall, K.; Barth, B. Posterior reversible encephalopathy syndrome in the emergency department: A single center retrospective study. Am. J. Emerg. Med. 2021, 45, 61–64. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country/# | Sex | Age (Y) | MofO | Genotype | ANC (×109/L) | HF | SF/EF at DGN (%) | LVEDD z-Score at DGN | LV Mass z-Score at DGN | LVNC | SGA | Age of Walking (M) | 3-MGCA | Informative CL Profile |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| France/1 | F | 0.09 | CMP | Del exon 1-5 | 0.85 | Yes | 9/24 | 3.1 | 4.6 | Yes | Yes | 24 | No | Yes |
| France/2 | M | 0.01 | CMP | Exon 2/c.143delinsGG/p.Glu48fsX | 1.93 | Yes | 20/38 | 4.5 | −0.4 | Yes | No | 24 | No | Yes |
| France/3 | M | 0.07 | INF | Exon 3/c.280C > A/p.Arg94Ser | 0.98 | Yes | N.A. | 0.7 | 1.4 | No | No | 20 | Yes | Yes |
| France/4 | M | 1.37 | CMP | Exon 3/c.281G > A/p.Arg94His | 2.9 | Yes | 19/30 | 13 | N.A. | No | No | N.A. | Yes | Yes |
| France/5 | M | 0.08 | CMP | Exon 4/c.356T > G/p.Val119Gly | 1.79 | Yes | 25/58 | 2.8 | N.A. | Yes | No | N.A. | Yes | Yes |
| France/6 | M | 0.69 | INF | Exon 6/c.478A > T/p. Lys160X | 0.3 | No | 34/70 | −0.7 | −0.9 | No | No | 18 | Yes | Yes |
| France/7 | M | 0.05 | CMP | Del exon 6-11 | 0.77 | Yes | 14/N.A. | N.A. | N.A. | No | Yes | N.A. | Yes | Yes |
| France/8 | M | 0.11 | CMP | Del exon 6-11 | 0.61 | Yes | 31/61 | 6.6 | N.A. | No | Yes | 18 | Yes | N.A. |
| France/9 | M | 0.13 | INF | Exon 8/c.589G > A/p.Gly197Arg | 1 | Yes | 10/N.A. | 6.6 | N.A. | No | No | N.A. | N.A. | Yes |
| France/10 | M | IU | CMP | Exon 8/c.589G > T/p.Gly197Trp | 0.52 | Yes | 12.3/23.8 | 7.5 | 6.4 | No | Yes | N.A. | Yes | Yes |
| France/11 | M | IU | CMP | Exon 8/c.646G > A/p.Gly216Arg | 13.63 | Yes | 25/N.A. | 1.3 | N.A. | No | Yes | N.A. | No | Yes |
| France/12 | M | IU | CMP | N.T. | 0.66 | Yes | 20/N.A. | 1.3 | N.A. | No | No | N.A. | N.A. | N.A. |
| France/13 | M | 0 | CMP | Exon 8/c.646G > A/p.Gly216Arg | N.A. | Yes | N.A. | N.A. | N.A. | No | No | N.A. | N.A. | N.A. |
| France/14 | M | 0.1 | INF | N.T. | 0 | No | N.A. | N.A. | N.A. | No | No | N.A. | N.A. | N.A. |
| France/15 | M | 0.71 | CMP | Del exon 8-9 | 2.5 | Yes | 13.7/25.6 | 12.7 | 7.8 | No | No | N.A. | N.A. | Yes |
| France/16 | M | 0.17 | CMP | Exon 9/c.659_660dupGTCC/p.Leu221fsX | 0.7 | Yes | 16/35 | 7.4 | 2 | No | No | N.A. | No | Yes |
| France/17 | M | 0 | CMP | Exon 9/c.659_660dupGTCC/p.Leu221fsX | 1.47 | Yes | 30/N.A. | 3.3 | 8.6 | Yes | No | N.A. | No | Yes |
| France/18 | M | 1.7 | FTT | Intron 9/c.700-1G > A/p. | 0.72 | Yes | 8.5/16.3 | 8.4 | 3.5 | Yes | Yes | N.A. | No | N.A. |
| France/19 | M | 0 | Low Glu | N.D. | 0.97 | Yes | 12.8/28.3 | 10.2 | 4.3 | Yes | Yes | 21 | Yes | Yes |
| France/20 | M | 0 | CMP | N.T. | 2.88 | Yes | N.A. | N.A. | N.A. | No | No | N.A. | N.A. | N.A. |
| France/21 | M | 0 | CMP | Intron 10/c. 778-1G > T | 3.82 | Yes | 16/36.1 | 1.9 | 3 | Yes | No | 18 | Yes | Yes |
| France/22 | M | 0 | CMP | Intron 10/c. 778-1G > T | 4.28 | Yes | N.A. | N.A. | N.A. | Yes | No | 12 | Yes | Yes |
| China/1 | M | 0.21 | INF | c.527A > G (p.H176R) | N.A. | No | 22.1/45.6 | 5.7 | N.A. | Yes | N/A | N.A. | Yes | N.A. |
| China/2 | M | 0.21 | HF | c.527A > G (p.H176R) | N.A. | Yes | 16.7/36.2 | 3.8 | N.A. | Yes | N/A | N.A. | Yes | N.A. |
| China/3 | M | 0.5 | DMT | c.367C > T (p.R123X) | N.A. | No | 19.1/40.1 | 3.3 | N.A. | Yes | N/A | N.A. | Yes | N.A. |
| China/4 | M | 0.54 | INF | c.710_711delTG (p.V237AfsX73) | N.A. | No | 17.3/36.8 | 5.3 | N.A. | Yes | N/A | N.A. | Yes | N.A. |
| China/5 | M | 0.08 | INF | c.134_136delinsCC (p.H45PfsX38) | N.A. | No | 18.9/40.1 | 5.7 | N.A. | Yes | N/A | N.A. | No | N.A. |
| China/6 | M | 0.12 | INF | N.D. | N.A. | No | 20.0/43.0 | 4 | N.A. | Yes | N/A | N.A. | N.D. | N.A. |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sergi, C.M. Barth Syndrome: TAFAZZIN Gene, Cardiologic Aspects, and Mitochondrial Studies—A Comprehensive Narrative Review. Genes 2025, 16, 465. https://doi.org/10.3390/genes16040465
Sergi CM. Barth Syndrome: TAFAZZIN Gene, Cardiologic Aspects, and Mitochondrial Studies—A Comprehensive Narrative Review. Genes. 2025; 16(4):465. https://doi.org/10.3390/genes16040465
Chicago/Turabian StyleSergi, Consolato M. 2025. "Barth Syndrome: TAFAZZIN Gene, Cardiologic Aspects, and Mitochondrial Studies—A Comprehensive Narrative Review" Genes 16, no. 4: 465. https://doi.org/10.3390/genes16040465
APA StyleSergi, C. M. (2025). Barth Syndrome: TAFAZZIN Gene, Cardiologic Aspects, and Mitochondrial Studies—A Comprehensive Narrative Review. Genes, 16(4), 465. https://doi.org/10.3390/genes16040465