Maternal Uniparental Isodisomy of Chromosome 6: A Novel Case of Teratoma and Autism Spectrum Disorder with a Diagnostic and Management Framework


 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
3. Results
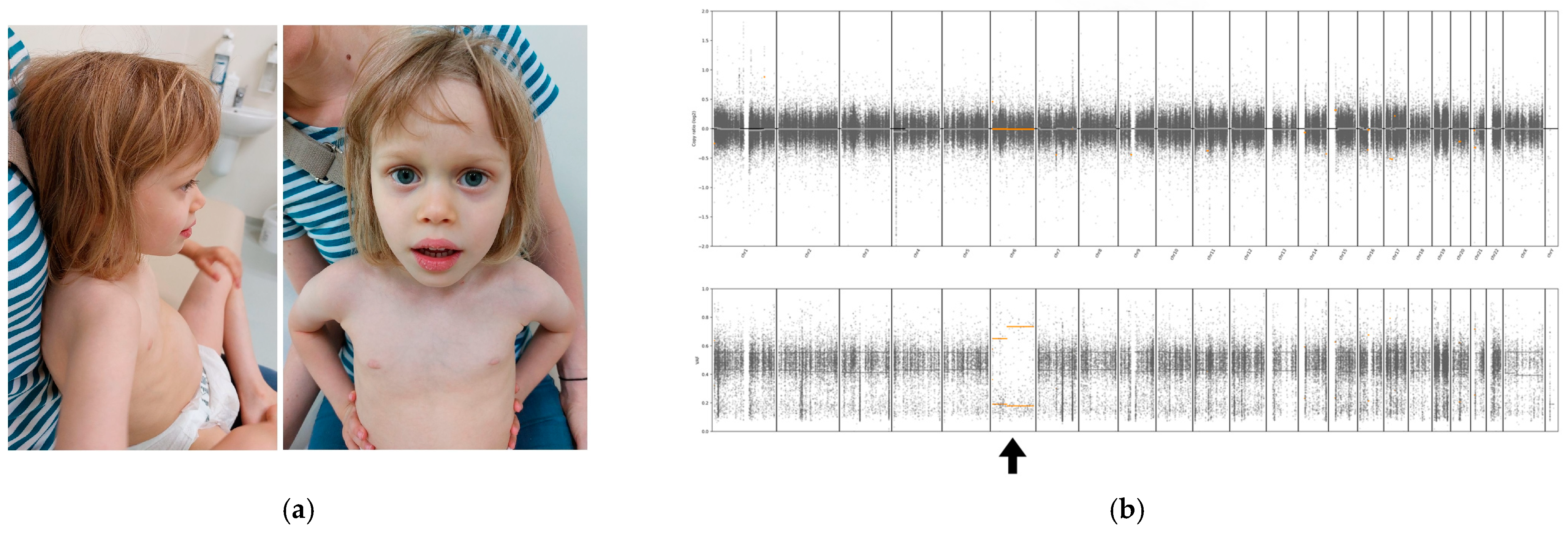
3.1. Case Report
3.2. Genetic Analysis
4. Discussion
4.1. Literature Review of UPD(6) Cases
4.2. Possible Underlying Molecular/Cytogenetic Mechanisms of UPD6
4.3. Neoplasms in Uniparental Disomy
4.4. Diagnosis and Management of UPD6 Cases
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| UPD | Uniparental disomy |
| UPD(6)mat | Maternal uniparental disomy of chromosome 6 |
| IUGR | Intrauterine growth restriction |
| ASD | Autism spectrum disorder |
| WES | Whole exome sequencing |
| Trio WES | Whole exome sequencing for the proband and both parents |
| MRI | Magnetic resonance imaging |
| EEG | Electroencephalography |
| ASRS | Autism Spectrum Rating Scales |
| LOH | Loss of heterozygosity |
| CPM | Confined placental mosaicism |
| CMA | Chromosomal microarray |
| MSA | Microsatellite analysis |
| MS-MLPA | Methylation-Specific Multiplex Ligation-Dependent Probe Amplification |
| NGS | Next-generation sequencing |
| TNDM | Transient neonatal diabetes mellitus |
| SNP | Single nucleotide polymorphism |
References
- Liehr, T. Uniparental disomy is a chromosomic disorder in the first place. Mol. Cytogenet. 2022, 15, 5. [Google Scholar]
- Engel, E. A new genetic concept: Uniparental disomy and its potential effect, isodisomy. Am. J. Med. Genet. 1980, 6, 137–143. [Google Scholar]
- Spence, J.; Perciaccante, R.; Greig, G.; Willard, H.; Ledbetter, D.; Hejtmancik, J.; Pollack, M.S.; Obrien, W.; Beaudet, A. Uniparental disomy as a mechanism for human genetic disease. Am. J. Hum. Genet. 1988, 42, 217–226. [Google Scholar]
- Nakka, P.; Smith, S.P.; O’donnell-Luria, A.H.; McManus, K.F.; Mountain, J.L.; Ramachandran, S.; Sathirapongsasuti, J.F.; Agee, M.; Auton, A.; Bell, R.K.; et al. Characterization of Prevalence and Health Consequences of Uniparental Disomy in Four Million Individuals from the General Population. Am. J. Hum. Genet. 2019, 105, 921–932. [Google Scholar] [PubMed]
- Del Gaudio, D.; Shinawi, M.; Astbury, C.; Tayeh, M.K.; Deak, K.L.; Raca, G. Diagnostic testing for uniparental disomy: A points to consider statement from the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 1133–1141. [Google Scholar] [PubMed]
- Temple, I.K.; Mackay, D.J.G. Diabetes Mellitus, 6q24-Related Transient Neonatal. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Gargano, M.A.; Matentzoglu, N.; Coleman, B.; Addo-Lartey, E.B.; Anagnostopoulos, A.V.; Anderton, J.; Avillach, P.; Bagley, A.M.; Bakštein, E.; Balhoff, J.P.; et al. The Human Phenotype Ontology in 2024: Phenotypes around the world. Nucleic Acids Res. 2024, 52, D1333–D1346. [Google Scholar] [PubMed]
- Fenton, T.R.; Kim, J.H. A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatr. 2013, 13, 59. [Google Scholar]
- Bione, S.; Sala, C.; Manzini, C.; Arrigo, G.; Zuffardi, O.; Banfi, S.; Borsani, G.; Jonveaux, P.; Philippe, C.; Zuccotti, M.; et al. A human homologue of the Drosophila melanogaster diaphanous gene is disrupted in a patient with premature ovarian failure: Evidence for conserved function in oogenesis and implications for human sterility. Am. J. Hum. Genet. 1998, 62, 533–541. [Google Scholar]
- Eggermann, T.; Oehl-Jaschkowitz, B.; Dicks, S.; Thomas, W.; Kanber, D.; Albrecht, B.; Begemann, M.; Kurth, I.; Beygo, J.; Buiting, K. The maternal uniparental disomy of chromosome 6 (upd(6)mat) “phenotype”: Result of placental trisomy 6 mosaicism? Mol. Genet. Genom. Med. 2017, 5, 668–677. [Google Scholar]
- Poke, G.; Doody, M.; Prado, J.; Gattas, M. Segmental Maternal UPD6 with Prenatal Growth Restriction. Mol. Syndromol. 2013, 3, 270–273. [Google Scholar]
- Docherty, L.E.; Kabwama, S.; Lehmann, A.; Hawke, E.; Harrison, L.; Flanagan, S.E.; Ellard, S.; Hattersley, A.T.; Shield, J.P.H.; Ennis, S.; et al. Clinical presentation of 6q24 transient neonatal diabetes mellitus (6q24 TNDM) and genotype-phenotype correlation in an international cohort of patients. Diabetologia 2013, 56, 758–762. [Google Scholar]
- Varrault, A.; Gueydan, C.; Delalbre, A.; Bellmann, A.; Houssami, S.; Aknin, C.; Severac, D.; Chotard, L.; Kahli, M.; Le Digarcher, A.; et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell 2006, 11, 711–722. [Google Scholar] [PubMed]
- Court, F.; Tayama, C.; Romanelli, V.; Martin-Trujillo, A.; Iglesias-Platas, I.; Okamura, K.; Sugahara, N.; Simón, C.; Moore, H.; Harness, J.V.; et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014, 24, 554–569. [Google Scholar]
- Jadhav, B.; Monajemi, R.; Gagalova, K.K.; Ho, D.; Draisma, H.H.; van de Wiel, M.A.; Franke, L.; Heijmans, B.T.; van Meurs, J.; Jansen, R.; et al. RNA-Seq in 296 phased trios provides a high-resolution map of genomic imprinting. BMC Biol. 2019, 17, 50. [Google Scholar]
- Wan, C.; Ma, H.; Liu, J.; Liu, F.; Liu, J.; Dong, G.; Zeng, X.; Li, D.; Yu, Z.; Wang, X.; et al. Quantitative relationships of FAM50B and PTCHD3 methylation with reduced intelligence quotients in school aged children exposed to lead: Evidence from epidemiological and in vitro studies. Sci. Total Environ. 2024, 907, 167976. [Google Scholar] [PubMed]
- Makishima, H.; Maciejewski, J.P. Pathogenesis and consequences of uniparental disomy in cancer. Clin. Cancer Res. 2011, 17, 3913–3923. [Google Scholar] [CrossRef]
- Barksdale, E.M., Jr.; Obokhare, I. Teratomas in infants and children. Curr. Opin Pediatr. 2009, 21, 344–349. [Google Scholar] [PubMed]
- Kearney, H.M.; Kearney, J.B.; Conlin, L.K. Diagnostic implications of excessive homozygosity detected by SNP-Based microarrays: Consanguinity, uniparental disomy, and recessive single-gene mutations. Clin. Lab. Med. 2011, 31, 595–613. [Google Scholar] [CrossRef]
- Hoppman, N.; Rumilla, K.; Lauer, E.; Kearney, H.; Thorland, E. Patterns of homozygosity in patients with uniparental disomy: Detection rate and suggested reporting thresholds for SNP microarrays. Genet. Med. 2018, 20, 1522–1527. [Google Scholar]
- Bilo, L.; Ochoa, E.; Lee, S.; Dey, D.; Kurth, I.; Kraft, F.; Rodger, F.; Docquier, F.; Toribio, A.; Bottolo, L.; et al. Molecular characterisation of 36 multilocus imprinting disturbance (MLID) patients: A comprehensive approach. Clin. Epigenetics 2023, 15, 35. [Google Scholar] [CrossRef]
- van den Berg-Loonen, E.M.; Savelkoul, P.; van Hooff, H.; van Eede, P.; Riesewijk, A.; Geraedts, J. Uniparental maternal disomy 6 in a renal transplant patient. Hum. Immunol. 1996, 45, 46–51. [Google Scholar] [CrossRef]
- Spiro, R.P.; Christian, S.L.; Ledbetter, D.H.; New, M.I.; Wilson, R.C.; Roizen, N.; Rosenfield, R.L. Intrauterine growth retardation associated with maternal uniparental disomy for chromosome 6 unmasked by congenital adrenal hyperplasia. Pediatr Res. 1999, 46, 510–513. [Google Scholar] [PubMed]
- Cockwell, A.E.; Baker, S.J.; Connarty, M.; Moore, I.E.; Crolla, J.A. Mosaic trisomy 6 and maternal uniparental disomy 6 in a 23-week gestation fetus with atrioventricular septal defect. Am. J. Med. Genet. 2006, 140, 624–627. [Google Scholar] [CrossRef]
- Parker, E.A.; Hovanes, K.; Germak, J.; Porter, F.; Merke, D.P. Maternal 21-hydroxylase deficiency and uniparental isodisomy of chromosome 6 and X results in a child with 21-hydroxylase deficiency and Klinefelter syndrome. Am J Med Genet A. 2006, 140, 2236–2240. [Google Scholar] [CrossRef] [PubMed]
- Gümüş, H.; Ghesquiere, S.; Per, H.; Kondolot, M.; Ichida, K.; Poyrazoğlu, G.; Kumandaş, S.; Engelen, J.; Dundar, M.; Cağlayan, A.O. Maternal uniparental isodisomy is responsible for serious molybdenum cofactor deficiency. Dev. Med. Child Neurol. 2010, 52, 868–872. [Google Scholar] [PubMed]
- Salahshourifar, I.; Halim, A.S.; Sulaiman, W.A.; Zilfalil, B.A. Maternal uniparental heterodisomy of chromosome 6 in a boy with an isolated cleft lip and palate. Am. J. Med. Genet A 2010, 152, 1818–1821. [Google Scholar] [CrossRef]
- Sasaki, K.; Okamoto, N.; Kosaki, K.; Yorifuji, T.; Shimokawa, O.; Mishima, H.; Yoshiura, K.I.; Harada, N. Maternal uniparental isodisomy and heterodisomy on chromosome 6 encompassing a CUL7 gene mutation causing 3M syndrome. Clin. Genet. 2011, 80, 478–483. [Google Scholar] [CrossRef]
- Begemann, M.; Spengler, S.; Gogiel, M.; Grasshoff, U.; Bonin, M.; Betz, R.C.; Dufke, A.; Spier, I.; Eggermann, T. Clinical significance of copy number variations in the 11p15.5 imprinting control regions: New cases and review of the literature. J. Med. Genet. 2012, 49, 547–553. [Google Scholar]
- Roosing, S.; van den Born, L.I.; Hoyng, C.B.; Thiadens, A.A.; de Baere, E.; Collin, R.W.; Koenekoop, R.K.; Leroy, B.P.; van Moll-Ramirez, N.; Venselaar, H. Maternal uniparental isodisomy of chromosome 6 reveals a TULP1 mutation as a novel cause of cone dysfunction. Ophthalmology 2013, 120, 1239–1246. [Google Scholar]
- Takimoto, T.; Takada, H.; Ishimura, M.; Kirino, M.; Hata, K.; Ohara, O.; Hara, T. Wiskott-Aldrich Syndrome in a Girl Caused by Heterozygous WASP Mutation and Extremely Skewed X-Chromosome Inactivation: A Novel Association with Maternal Uniparental Isodisomy 6. Neonatology 2015, 107, 185–190. [Google Scholar]
- Lazier, J.; Martin, N.; Stavropoulos, J.D.; Chitayat, D. Maternal uniparental disomy for chromosome 6 in a patient with IUGR, ambiguous genitalia, and persistent mullerian structures. American Journal of Medical Genetics, Part A. 2016, 170, 3227–3230. [Google Scholar] [PubMed]
- Leung, W.C.; Lau, W.L.; Lo, T.K.; Lau, T.K.; Lam, Y.Y.; Kan, A.; Chan, K.; Lau, E.T.; Tang, M.H. Two IUGR foetuses with maternal uniparental disomy of chromosome 6 or UPD(6)mat. J. Obstet. Gynaecol. 2017, 37, 113–115. [Google Scholar]
- Kerr, E.R.; Stuhlmiller, G.M.; Maha, G.C.; Ladd, M.A.; Mikhail, F.M.; Koester, R.P.; Hurst, A.C. Maternal uniparental isodisomy for chromosome 6 discovered by paternity testing: A case report. Mol. Cytogenet. 2018, 11, 60. [Google Scholar] [PubMed]
- Souzeau, E.; Thompson, J.A.; McLaren, T.L.; De Roach, J.N.; Barnett, C.P.; Lamey, T.M.; Craig, J.E. Maternal uniparental isodisomy of chromosome 6 unmasks a novel variant in TULP1 in a patient with early onset retinal dystrophy. Mol. Vis. 2018, 24, 478–484. [Google Scholar]
- Zhang, P.; Ying, W.; Wu, B.; Liu, R.; Wang, H.; Wang, X.; Lu, Y. Complete IFN-γR1 Deficiency in a Boy Due to UPD(6)mat with IFNGR1 Novel Splicing Variant. J. Clin. Immunol. 2021, 41, 834–836. [Google Scholar]
- Jiang, Y.; Xiao, Y.X.; Xiong, J.J.; Zhang, V.W.; Dong, C.; Xu, L.; Liu, F. Maternal uniparental disomy for chromosome 6 in 2 prenatal cases with IUGR: Case report and literature review. Mol. Cytogenet. 2024, 17, 1. [Google Scholar] [PubMed]
- Li, J.W.; Qian, Y.J.; Mao, S.J.; Chao, Y.Q.; Qin, Y.F.; Hu, C.X.; Li, Z.L.; Zou, C.C. Clinical features associated with maternal uniparental disomy for chromosome 6. Mol. Cytogenet. 2024, 17, 18. [Google Scholar]
- Harris, L.K.; Pantham, P.; Yong, H.E.J.; Pratt, A.; Borg, A.J.; Crocker, I.; Westwood, M.; Aplin, J.; Kalionis, B.; Murthi, P. The role of insulin-like growth factor 2 receptor-mediated homeobox gene expression in human placental apoptosis, and its implications in idiopathic fetal growth restriction. Mol. Hum. Reprod. 2019, 25, 572–585. [Google Scholar]
- Bradfield, J.P.; Qu, H.Q.; Wang, K.; Zhang, H.; Sleiman, P.M.; Kim, C.E.; Mentch, F.D.; Qiu, H.; Glessner, J.T.; Thomas, K.A.; et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011, 7, e1002293. [Google Scholar]
- Xu, Y.; Goodyer, C.G.; Deal, C.; Polychronakos, C. Functional polymorphism in the parental imprinting of the human IGF2R gene. Biochem. Biophys. Res. Commun. 1993, 197, 747–754. [Google Scholar]
- Mozaffari, S.V.; Stein, M.M.; Magnaye, K.M.; Nicolae, D.L.; Ober, C. Parent of origin gene expression in a founder population identifies two new candidate imprinted genes at known imprinted regions. PLoS ONE 2018, 13, e0203906. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| UPD Criteria | Our Case |
|---|---|
| Gender and age of patient | Female; 4 years and 10 months |
| Prenatal | Polyhydramnios [HP:0001561]; IUGR [HP:0001511] |
| Clinical phenotype | Delayed speech and language development [HP:0000750]; Autism spectrum disorder [HP:0000729]; Triangular face [HP:0000325]; Prominent forehead [HP:0011220]; Full lips [HP:0012471]; Reduced subcutaneous adipose tissue [HP:0003758]; Sacrococcygeal teratoma [HP:0030736]; Sleep disturbance [HP:0002360]; Teeth grinding [HP:0003763]; Feeding difficulties [HP:0011968]; Auditory hypersensitivity [HP:5200060]; Genu valgum [HP:0002857]; Pes planus [HP:0001763]; Short attention span [HP:0000736] |
| Type of disomy | Maternal isodisomy |
| Affected chromosome | Whole chromosome 6 |
| Test performed | Whole exome sequencing (WES) in the proband, her mother, and her father (trio) |
| A mosaic present | No mosaic present (peripheral blood cells examined only) |
| System/Concern | Evaluation | Comment | Treatment (If Needed) | Surveillance [Frequency] |
|---|---|---|---|---|
| Pregnancy and Perinatal Period | Measurements of weight, length, head circumference at birth, and the age at birth | Note IUGR, low birth weight, preterm labor, and any events during pregnancy | If any delayed growth signs are present, monitor further growth closely using appropriate percentile charts for preterm infants—Fenton [as needed] | |
| Constitutional | Measurements of weight, height, head circumference | Monitor further growth closely using appropriate percentile charts | Evaluate the need for recombinant human growth hormone (rhGH) therapy | Measurement of growth parameters on percentile charts, monitor weight and height increments [at each visit] |
| Development, Behavior, and Neurological | Assessment of development and speech-language skills | Brain MRI if significant developmental delay is observed; Monitor for any features characteristic of Autism spectrum disorder; Monitor neurobehavioral patterns of the child | Neuropsychological therapy, cognitive therapy, speech therapy; Evaluate the need for special education institutions | Monitor developmental progress and educational needs [at each visit] |
| Musculoskeletal, Orthopedic, and Motor Skills | Evaluation of muscle tension, gross motor skills; Assessment of knee and foot alignment | Monitor for conditions such as pes planus and genu valgum | Physical therapy and orthopedic intervention as needed | Assessment of reaching motor milestones [at each visit] |
| Feeding | Evaluation of feeding effectiveness and dysphagia | Ensure adequate nutrition and assess for feeding difficulties | Nutritional support and feeding therapy | Monitor [at each visit] |
| Neoplasia | Abdominal ultrasound | Screen for any masses or tumors | Oncologic care as needed | |
| Facial Features and Skin | Assessment of facial features, subcutaneous tissue, skin characteristics | Evaluation for facial dysmorphia, facial clefts, lack of subcutaneous fat tissue | ||
| Renal | Renal function tests and renal ultrasound | Monitor for renal insufficiency and structural anomalies | Nephrologic care as needed | |
| Adrenal | Screening for congenital adrenal hyperplasia (CAH) | Hormone replacement therapy if indicated | ||
| Cardiovascular | Echocardiography | To assess heart morphology and for patent ductus arteriosus | Cardiac care as needed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Świeca, A.; Franaszczyk, M.; Maryniak, A.; Lipiński, P.; Płoski, R.; Szczałuba, K. Maternal Uniparental Isodisomy of Chromosome 6: A Novel Case of Teratoma and Autism Spectrum Disorder with a Diagnostic and Management Framework. Genes 2025, 16, 434. https://doi.org/10.3390/genes16040434
Świeca A, Franaszczyk M, Maryniak A, Lipiński P, Płoski R, Szczałuba K. Maternal Uniparental Isodisomy of Chromosome 6: A Novel Case of Teratoma and Autism Spectrum Disorder with a Diagnostic and Management Framework. Genes. 2025; 16(4):434. https://doi.org/10.3390/genes16040434
Chicago/Turabian StyleŚwieca, Aleksandra, Maria Franaszczyk, Agnieszka Maryniak, Patryk Lipiński, Rafał Płoski, and Krzysztof Szczałuba. 2025. "Maternal Uniparental Isodisomy of Chromosome 6: A Novel Case of Teratoma and Autism Spectrum Disorder with a Diagnostic and Management Framework" Genes 16, no. 4: 434. https://doi.org/10.3390/genes16040434
APA StyleŚwieca, A., Franaszczyk, M., Maryniak, A., Lipiński, P., Płoski, R., & Szczałuba, K. (2025). Maternal Uniparental Isodisomy of Chromosome 6: A Novel Case of Teratoma and Autism Spectrum Disorder with a Diagnostic and Management Framework. Genes, 16(4), 434. https://doi.org/10.3390/genes16040434