
The Interconnection Between UbcH10, p53, and EGFR in Lung Cancer Cells and Their Involvement in Treatment Response

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Transfections
2.2. Proliferative Studies
2.3. Protein Extraction, Western Blotting, and Immunoprecipitation
2.4. Selection of NSCLC Tissue Specimens
2.5. Immunohistochemistry
2.6. Statistical Methods
3. Results
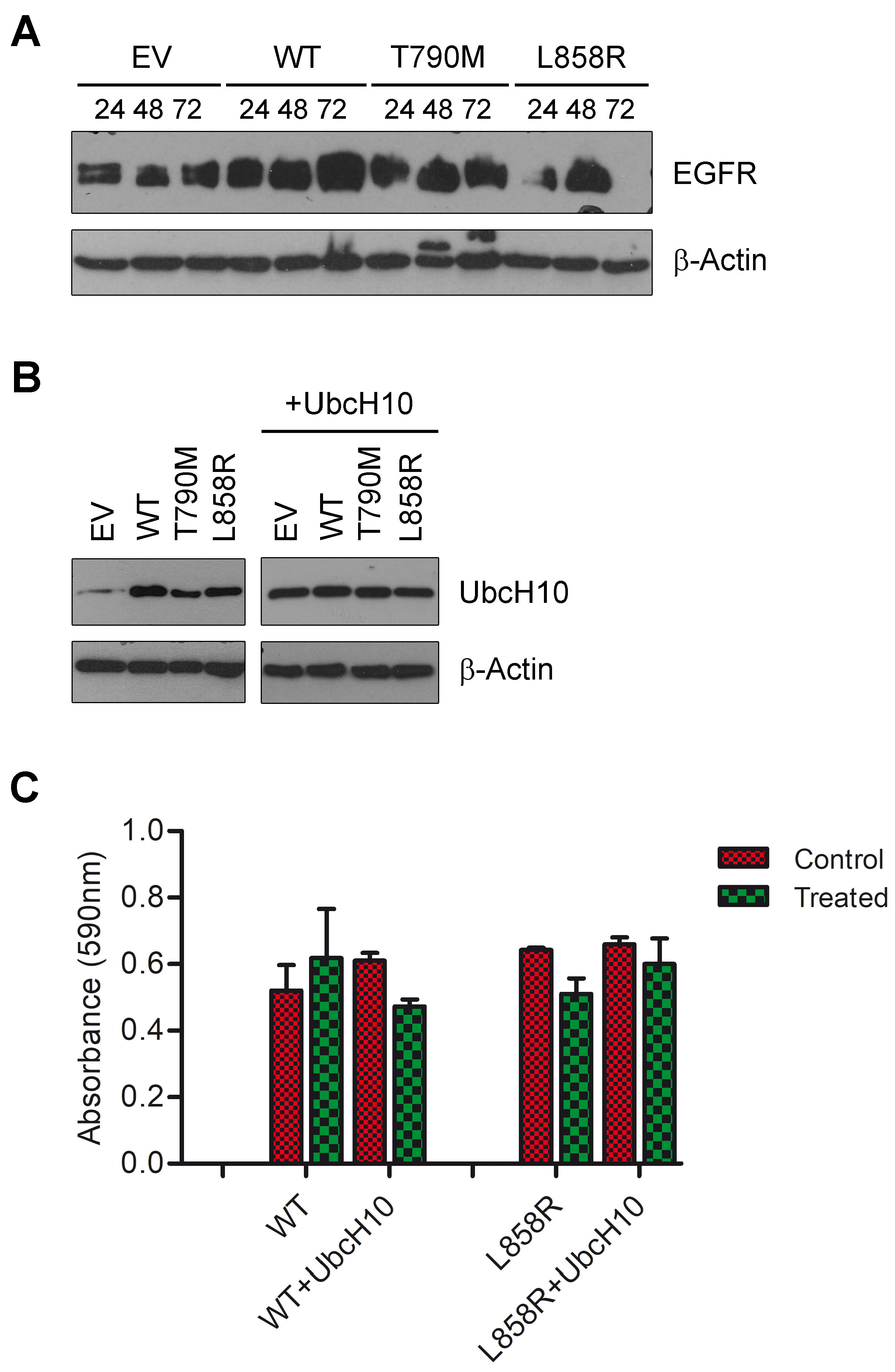
3.1. UbcH10 Modulates Drug Sensitivity of Lung Cancer Cells Carrying Mutant EGFR
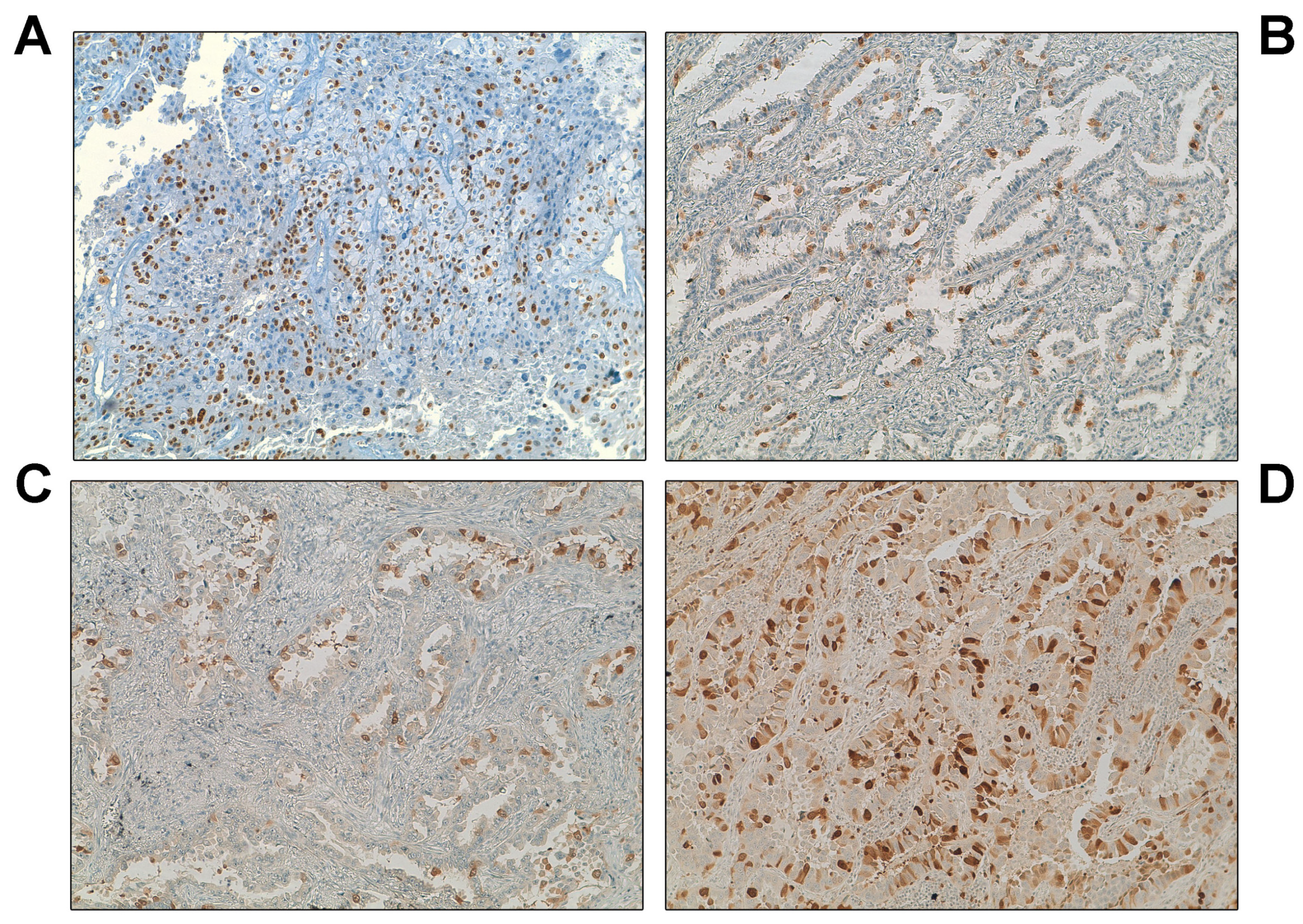
3.2. Overexpression of UbcH10 Is Associated with Resistance in NSCLC Patients
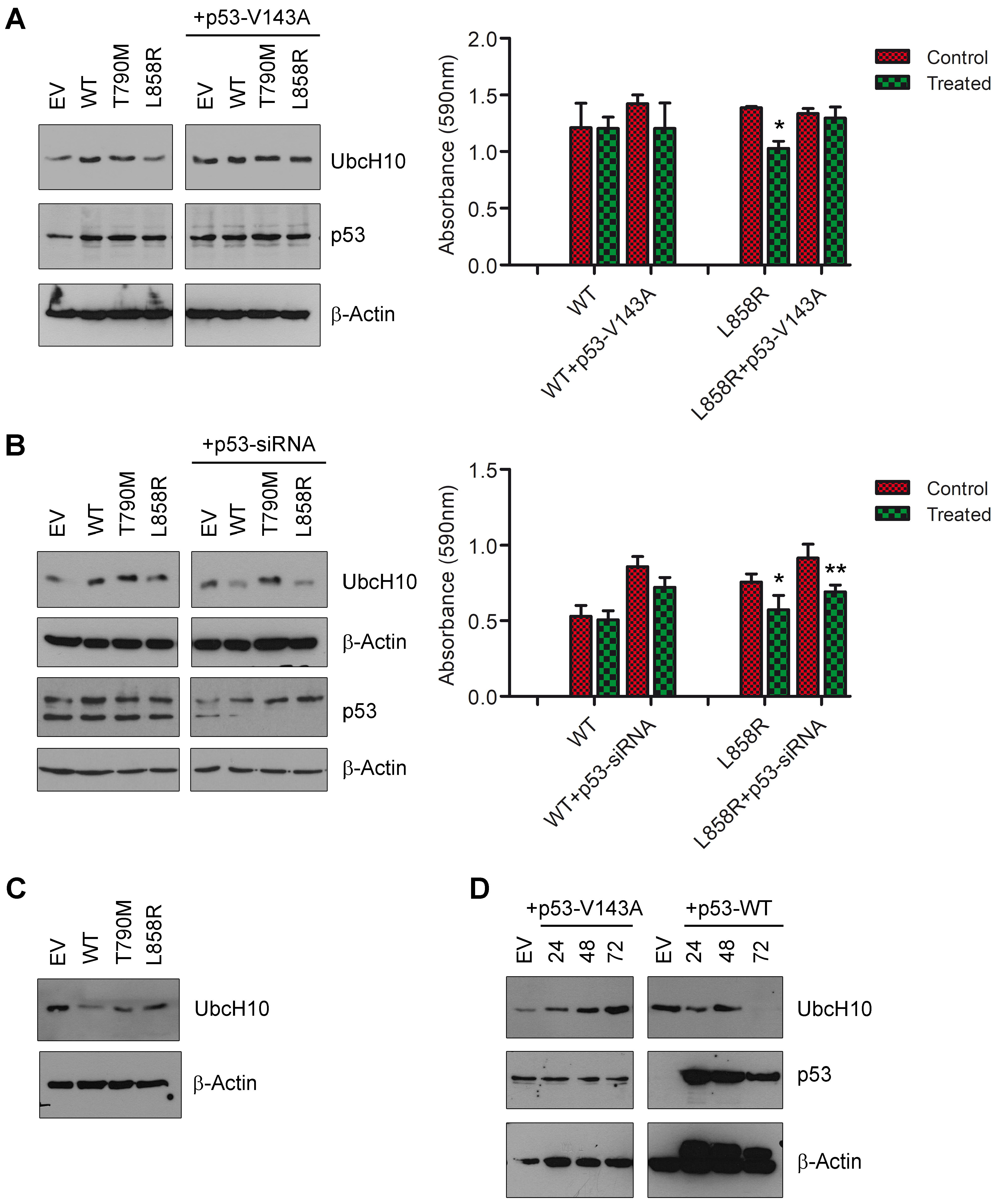
3.3. P53 May Influence UbcH10 Expression in Lung Cancer Cells
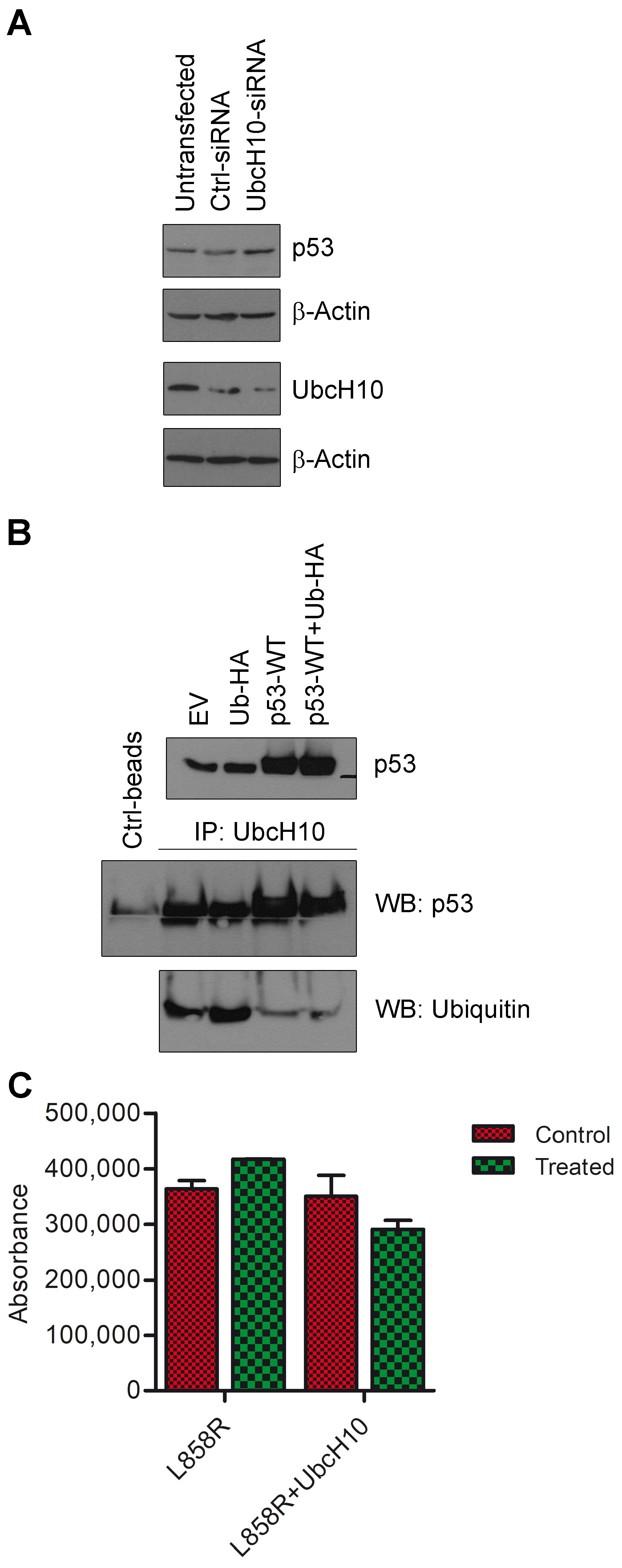
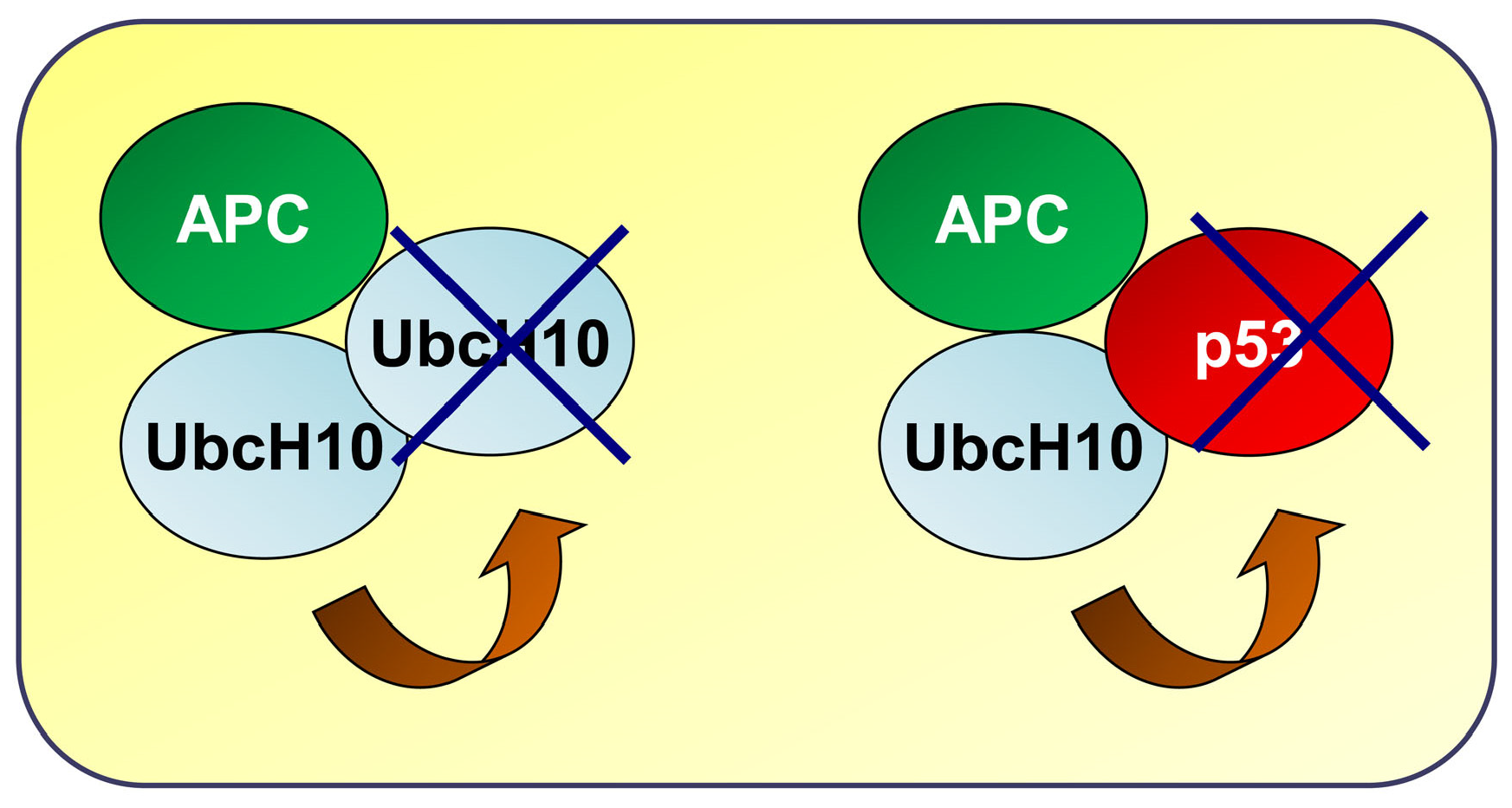
3.4. Potential Self-Regulatory Relationship Between UbcH10 and p53
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NSCLC | Non-small cell lung cancer |
| SCLC | Small cell lung cancer |
| AD | Adenocarcinoma |
| TKI | Tyrosine kinase inhibitor |
| SD | Stable disease |
| PR | Partial response |
| CR | Complete response |
| PD | Progressing disease |
| APC | Anaphase-promoting complex |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Barr, T.; Ma, S.; Li, Z.; Yu, J. Recent Advances and Remaining Challenges in Lung Cancer Therapy. Chin. Med. J. 2024, 137, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Remon, J.; Hellmann, M.D. First-Line Immunotherapy for Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 586–597. [Google Scholar]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR -Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung, Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Piotrowska, Z.; Hata, A.N. Resistance to First-Line Osimertinib in EGFR-Mutant NSCLC: Tissue Is the Issue. Clin. Cancer Res. 2020, 26, 2441–2443. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung Cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Cacciola, N.A.; Calabrese, C.; Malapelle, U.; Pellino, G.; De Stefano, A.; Sepe, R.; Sgariglia, R.; Quintavalle, C.; Federico, A.; Bianco, A.; et al. UbcH10 Expression Can Predict Prognosis and Sensitivity to the Antineoplastic Treatment for Colorectal Cancer Patients. Mol. Carcinog. 2016, 55, 793–807. [Google Scholar] [CrossRef]
- Fujita, T.; Ikeda, H.; Taira, N.; Hatoh, S.; Naito, M.; Doihara, H. Overexpression of UbcH10 Alternates the Cell Cycle Profile and Accelerate the Tumor Proliferation in Colon Cancer. BMC Cancer 2009, 9, 87. [Google Scholar] [CrossRef]
- Presta, I.; Novellino, F.; Donato, A.; La Torre, D.; Palleria, C.; Russo, E.; Malara, N.; Donato, G. Ubch10 a Major Actor in Cancerogenesis and a Potential Tool for Diagnosis and Therapy. Int. J. Mol. Sci. 2020, 21, 2041. [Google Scholar] [CrossRef]
- Zhao, M.; Li, J.; Wang, R.; Mi, L.; Gu, Y.; Chen, R.; Li, Y.; Shi, W.; Zhang, Y. Ubiquitination-Binding Enzyme 2C Is Associated with Cancer Development and Prognosis and Is a Potential Therapeutic Target. OncoTargets Ther. 2024, 17, 1159–1171. [Google Scholar]
- Van Ree, J.H.; Jeganathan, K.B.; Malureanu, L.; Van Deursen, J.M. Overexpression of the E2 Ubiquitin-Conjugating Enzyme UbcH10 Causes Chromosome Missegregation and Tumor Formation. J. Cell Biol. 2010, 188, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Ishibashi, Y.; Urashima, M.; Omura, N.; Nakada, K.; Nishikawa, K.; Shida, A.; Takada, K.; Kashiwagi, H.; Yanaga, K. High UBCH10 Protein Expression as a Marker of Poor Prognosis in Esophageal Squamous Cell Carcinoma. Anticancer Res. 2014, 34, 955–962. [Google Scholar] [PubMed]
- Li, H.; Yang, C.; Chen, K.; Sun, M. Expression Significance of Emi1, UBCH10 and CyclinB1 in Esophageal Squamous Cell Carcinoma. Pathol. Oncol. Res. 2023, 29, 1611081. [Google Scholar] [CrossRef]
- Pallante, P.; Malapelle, U.; Berlingieri, M.T.; Bellevicine, C.; Sepe, R.; Federico, A.; Rocco, D.; Galgani, M.; Chiariotti, L.; Sanchez-Cespedes, M.; et al. UbcH10 Overexpression in Human Lung Carcinomas and Its Correlation with EGFR and P53 Mutational Status. Eur. J. Cancer 2013, 49, 1117–1126. [Google Scholar] [CrossRef]
- Quintavalle, C.; Di Costanzo, S.; Zanca, C.; Tasset, I.; Fraldi, A.; Incoronato, M.; Mirabelli, P.; Monti, M.; Ballabio, A.; Pucci, P.; et al. Phosphorylation-Regulated Degradation of the Tumor-Suppressor Form of PED by Chaperone-Mediated Autophagy in Lung Cancer Cells. J. Cell. Physiol. 2014, 229, 1359–1368. [Google Scholar] [CrossRef]
- Lee, C.S.; Sharma, S.; Miao, E.; Mensah, C.; Sullivan, K.; Seetharamu, N. A Comprehensive Review of Contemporary Literature for Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer and Their Toxicity. Lung Cancer Targets Ther. 2020, 11, 73–103. [Google Scholar] [CrossRef]
- Huang, S.; Li, X. UBE2C Promotes LUAD Progression by Ubiquitin-Dependent Degradation of P53 to Inactivate the P53/P21 Signaling Pathway. Discov. Oncol. 2024, 15, 589. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, R.; Chi, S.; Zhang, W.; Xiao, C.; Zhou, X.; Zhao, Y.; Wang, H. UBE2C Is Upregulated by Estrogen and Promotes Epithelial-Mesenchymal Transition via P53 in Endometrial Cancer. Mol. Cancer Res. 2020, 18, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Wang, D.; Du, X.; Feng, X.; Zhu, X.; Wang, C. UBE2C Enhances Temozolomide Resistance by Regulating the Expression of P53 to Induce Aerobic Glycolysis in Glioma. Acta Biochim. Biophys. Sin. 2024, 56, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wu, M.; Bian, S.; Song, Q.; Xiao, M.; Huang, H.; You, L.; Zhang, J.; Zhang, J.; Cheng, C.; et al. DNA Primase Subunit 1 Deteriorated Progression of Hepatocellular Carcinoma by Activating AKT/MTOR Signaling and UBE2C-Mediated P53 Ubiquitination. Cell Biosci. 2021, 11, 42. [Google Scholar] [CrossRef]
- Rape, M.; Kirschner, M.W. Autonomous Regulation of the Anaphase-Promoting Complex Couples Mitosis to S-Phase Entry. Nature 2004, 432, 588–595. [Google Scholar] [CrossRef]
- Jiang, L.; Bao, Y.; Luo, C.; Hu, G.; Huang, C.; Ding, X.; Sun, K.; Lu, Y. Knockdown of Ubiquitin-Conjugating Enzyme E2C/UbcH10 Expression by RNA Interference Inhibits Glioma Cell Proliferation and Enhances Cell Apoptosis in Vitro. J. Cancer Res. Clin. Oncol. 2010, 136, 211–217. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does P53 Induce Apoptosis and How Does This Relate to P53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Kikuchi, S.; Nishimura, R.; Osako, T.; Okumura, Y.; Nishiyama, Y.; Toyozumi, Y.; Arima, N. Definition of P53 Overexpression and Its Association with the Clinicopathological Features in Luminal/HER2-Negative Breast Cancer. Anticancer Res. 2013, 33, 3891–3898. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample 1 | Histotype 1 | UbcH10 (% of Cells) | UbcH10 (Intensity) | Best Response 2 | H-Score 3 | H-Score (Mean) |
|---|---|---|---|---|---|---|
| W1 | AD | 60% | 3+ | n.a. | 180 | 96.7 |
| W2 | SCC | 10% | 1+ | n.a. | 10 | |
| W3 | NSCLC-n.o.s. | 50% | 2+ | n.a. | 100 | |
| M2 | AD | 5% | 2+ | CR | 10 | 12.5 |
| M5 | AD | 5% | 3+ | CR | 15 | |
| M4 | AD | 15% | 2+ | SD | 30 | 45 |
| M9 | AD | 30% | 3+ | SD | 90 | |
| M11 | AD | 5% | 3+ | SD | 15 | |
| M1 | AD | 10% | 2+ | PR | 20 | 61.7 |
| M3 | AD | 40% | 3+ | PR | 120 | |
| M8 | AD | 15% | 3+ | PR | 45 | |
| M10 | AD | 15% | 3+ | PD | 45 | 45 |
| M6 | AD | 5% | 3+ | n.e. | 15 | 22.5 |
| M7 | AD | 10% | 3+ | n.e. | 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quintavalle, C.; Malapelle, U.; De Martino, M.; Rocco, D.; Fusco, A.; Pepe, F.; Bellevicine, C.; Esposito, F.; Pallante, P. The Interconnection Between UbcH10, p53, and EGFR in Lung Cancer Cells and Their Involvement in Treatment Response. Genes 2025, 16, 404. https://doi.org/10.3390/genes16040404
Quintavalle C, Malapelle U, De Martino M, Rocco D, Fusco A, Pepe F, Bellevicine C, Esposito F, Pallante P. The Interconnection Between UbcH10, p53, and EGFR in Lung Cancer Cells and Their Involvement in Treatment Response. Genes. 2025; 16(4):404. https://doi.org/10.3390/genes16040404
Chicago/Turabian StyleQuintavalle, Cristina, Umberto Malapelle, Marco De Martino, Danilo Rocco, Alfredo Fusco, Francesco Pepe, Claudio Bellevicine, Francesco Esposito, and Pierlorenzo Pallante. 2025. "The Interconnection Between UbcH10, p53, and EGFR in Lung Cancer Cells and Their Involvement in Treatment Response" Genes 16, no. 4: 404. https://doi.org/10.3390/genes16040404
APA StyleQuintavalle, C., Malapelle, U., De Martino, M., Rocco, D., Fusco, A., Pepe, F., Bellevicine, C., Esposito, F., & Pallante, P. (2025). The Interconnection Between UbcH10, p53, and EGFR in Lung Cancer Cells and Their Involvement in Treatment Response. Genes, 16(4), 404. https://doi.org/10.3390/genes16040404