Abstract
Background/Objectives: To date, no information is available on the complete mitochondrial genome of the genus Dianema (Siluriformes: Callichthyidae), a callichthyid catfish. In this study, we report on two complete mitochondrial genome sequences of Dianema longibarbis Cope, 1872, and Dianema urostriatum Miranda Ribeiro, 1912, the only two recognized species within the genus Dianema. Methods: DNA sequencing was performed using the HiSeq platform to obtain their complete mitogenomes. To confirm phylogenetic distance, two phylogenetic trees were established using maximum-likelihood and Bayesian inference methods with all concatenated protein-coding sequences (PCGs) and two ribosomal RNA (rRNA) genes from the D. longibarbis and D. urostriatum mitogenomes, along with 32 mitogenomes retrieved from Siluriformes. Results: The complete mitogenomes of D. longibarbis and D. urostriatum are 16,493 and 16,495 base pairs in length, respectively. Their nucleotide compositions are 31.79% A, 27.53% T, 25.86% C, and 14.82% G for D. longibarbis, and 31.69% A, 27.04% T, 26.36% C, and 14.91% G for D. urostriatum. Both mitogenomes contain 13 PCGs, 22 transfer RNA (tRNA) genes, and two rRNA genes. Phylogenetic results based on all PCGs and two rRNAs genes confirm D. longibarbis as a sister species to D. urostriatum in the subfamily Callichthyinae. Conclusions: In contrast to the extensive mitochondrial studies on species in the Corydoradinae, species in the Callichthyinae have been largely understudied. This study provides valuable insights into genetic diversity and evolutionary complexity by presenting the first mitochondrial genome analysis of two Dianema species, a genus within the Callichthyinae.
1. Introduction
Siluriformes, comprising 35 families, 446 genera and 2867 species, represent approximately 10.8% of all fishes and are distributed worldwide [1,2,3]. The family Callichthyidae, which includes the highly commercialized ornamental Corydoras catfish, is found exclusively in the Neotropics [1,4]. Species in this family possess two rows of armor-like plates and are known for their ability to breathe oxygen using the intestines as a secondary respiratory system [5]. The Callichthyidae are divided into the subfamilies Corydoradinae and Callichthyinae. Of the 200 species within Callichthyidae, only 17 belong to the Callichthyinae [6]. Molecular and phenotypic classifications strongly support the monophyly of Callichthyinae, which consists of five genera: Callichthys, Dianema, Hoplosternum, Leptoplosternim, and Megalechis [7,8,9].
The genus Dianema in Callichthyinae consists of elongated, slender catfish, comprising only two species (Dianema longibarbis Cope, 1872 and Dianema urostriatum Miranda Ribeiro, 1912) [6]. Species of the genus Dianema are widely distributed in the neotropical central Amazon basin and are morphologically and behaviorally distinguished from other genera in Callichthyinae [7,8]. Unlike many Corydoradinae catfishes, Dianema species have long barbels that help them detect food. D. urostriatum is easily identified by the alternating white and black stripes on its caudal fin [10], whereas D. longibarbis is distinguished by the absence of a black stripe on its tail.
In general, the complete mitogenome, which consists of 13 PCGs, two rRNAs, 22 tRNAs, and a control region, is currently the most reliable molecular marker in evolutionary phylogenetics of fish due to its intrinsic features such as maternal inheritance and high base substitution rate [11,12,13]. Although more than 19 complete mitogenomes have been studied in the Corydoradinae, only one species, Hoplosternum littiorale, which has a partial mitogenome, has been studied in the Callichthyinae [14]. Therefore, previous phylogenies of the Callichthyinae were based on morphology or partial genes of mitogenomes [7,8,9,15]. Currently, most studies of complete mitochondrial genomes in the Callichthyidae have focused on the Corydorinae, and studies on the Callichthyinae are very scarce. For the first time, this study provides information on the complete mitogenomes of the two species comprising the genus Dianema, D. longibarbis and D. urostriatum, which will be invaluable in resolving the cryptic molecular phylogeny of the Callichthyinae.
2. Materials and Methods
2.1. Fish and DNA Extraction
Multiple specimens of D. longibarbis and D. urostriatum were acquired through the aquarium trade (AquaPro Trading, Namyangju, Republic of Korea). Photographs of the individuals used in the experiments are provided in the Supplementary Materials (Figure S1). Muscle tissue collected from each species was prepared for total genomic DNA isolation and subsequently cataloged at the Research Institute of Basic Sciences (Incheon National University, Incheon, Republic of Korea), with Specimen ID: 2024-Callichthyidae-03 and 04. Total genomic DNA was extracted with the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) based on the manufacturer’s protocols. The genomic DNA extracted was assessed for purity and concentration using a Nanodrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and was then stored at −20 °C until used for analysis.
2.2. DNA Sequencing, Assembly, and Gene Annotation
Next-generation sequencing was executed to obtain the complete mitogenomes of D. longibarbis and D. urostriatum. This was accomplished utilizing the HiSeq platform (150 bp; HiSeq X ten; Illumina, San Diego, CA, USA) based on previously established protocols [16]. To prepare for Illumina HiSeq sequencing, a fragment library was constructed using the TruSeq DNA Sample Preparation Kit (Illumina, San Diego, CA, USA) in accordance with the manufacturer’s suggestions at Macrogen, Inc. (Seoul, Republic of Korea). This entailed the random fragmentation of isolated total genomic DNA of each fish, followed by the ligation process with 5′ and 3′ adapters. Subsequently, the libraries underwent sequencing on the Illumina HiSeq platform, and the synthesized paired-end raw reads were tested through a stringent quality control procedure using FastQC version 0.11.9 [17]. After the demultiplexing process, only index pairs that matched were retrieved for further procedures. The raw read data underwent a meticulous quality trimming test, during which sequences for the 5′ and 3′ adapters, reads including low-quality sequences, reads with more than 10% unknown sequences, and reads containing ambiguous sequences were diligently removed using Trimmomatic [18], ultimately yielding a high-quality assembly. From an initial total of 4,560,049,755 and 4,800,833,497 raw reads for D. longibarbis and D. urostriatum, respectively, we obtained a final 30,259,360 and 31,853,978 filtered reads. To obtain circular contigs of the D. longibarbis and D. urostriatum mitogenomes, de novo assemblies were performed using different k-mers via NOVOplasty [19]. The average depth of coverage is provided in Figure S2. The resultant consensus sequence of the contig was annotated using MITOS2 [20]. The creation of mitochondrial genome maps for D. longibarbis and D. urostriatum were conducted through Proksee [21].
2.3. Mitogenome Analysis
The nucleotide composition and codon frequency and relative synonymous codon usage (RSCU) of both mitogenomes were calculated through MEGA 11 version 11.0.13 [22]. AT-skew and GC-skew for each mitogenome were analyzed with the equations of AT-skew = (A − T)/(A + T) and GC-skew = (G − C)/(G + C) for each fish, respectively. Prediction on the secondary structure for each tRNA was performed in the tRNASCAN webserver [23]. The control region sequences of 24 species belonging to Callichthyidae were obtained in the NCBI database. The tandem repeat sequences of the control region were predicted for each species using tandem repeat finder [24]. Nucleotide diversity (π) and nucleotide distances via the K2P model [25] for 13 PCGs of the 24 species of Callichthyidae in Table S1 were calculated using MEGA 11 version 11.0.13 [22]. The Ka/Ks values were analyzed with DnaSP version 6.12.03 [26].
2.4. Phylogenetic Analysis
To assess the phylogenetic relationships of D. longibarbis and D. urostriatum, maximum-likelihood (ML) and Bayesian inferences (BI) phylogenetic trees were constructed using concatenated amino acid coding sequence from all PCGs and two rRNA genes from the mitogenomes of D. longibarbis and D. urostriatum, with 34 mitogenomes retrieved from Siluriformes (24 species in Callichthyidae, seven species in Loricariidae, two species in Siluridae, and one species in Trichomycteridae,). The references for the GenBank accession number of each fish used in the phylogenetic analysis are appended in Supplementary Materials (Table S1). The 13 PCGs and two rRNA genes of each fish were obtained from the NCBI database. Sequences of the 13 PCGs and two rRNA genes were aligned through the L-INS-I algorithm with MAFFT version 7.490 [27]. Redundant gaps in each gene were trimmed using trimAl version 1.4 [28]. The modified sequences were then used to generate two matrices, one for the 13 PCGs and another for the combination of two rRNA + 13 PCGs, using SequenceMatrix version 1.8.1 and subsequently underwent conversion process to obtain nexus format [29]. To find the best substitution model for the sequences, the ModelFinder in IQ-TREE2 version 2.0.7 was used (Table S2) [30]. The ML-based phylogenetic result was established with 1000 replications of ultrafast bootstrapping with the optimal substitution model based on Bayesian Information Criterion (BIC) in IQ-TREE2 version 2.0.7 [31]. The BI-based phylogenetic tree was constructed using MrBayes program version 3.2.7, based on the best substitution model estimated through Akaike Information Criterion (AIC) [32]. Two independent Markov Chain Monte Carlo (MCMC) samplings of one million generations were applied using four chains and the tree was sampled every 100 generations. The effective sample size (ESS) was calculated with Tracer version 1.7.2, and all metrics were above 200, except for 25% burn-in [33]. Of the generated trees, the first 25% were removed as burn-in using LogCombiner version 2.7, and a consensus phylogenetic tree was constructed with TreeAnnotator version 2.7.6 [34]. The generated ML and BI trees were edited and visualized using Figtree version 1.4.4 [35].
3. Results
3.1. Mitogenome Structure and Base Composition
The complete mitogenomes of D. longibarbis and D. urostriatum are 16,493 bp and 16,495 bp long, respectively (GenBank accession no. PP737535 and PP790961) (Figure 1a,b and Table 1). In comparison to the mitogenomes of the other 20 species in the Callichthyidae, which range from 16,531 to 16,916 bp, these are among the shortest. Both mitogenomes contain a total of 13 PCGs, two rRNA genes (rrnS (12S) and rrnL (16S)), 22 tRNA genes, and one control region (Figure 1a,b and Table 1).
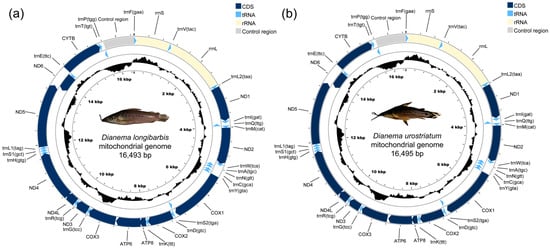
Figure 1.
The circular maps of the assembled mitogenomes of (a) D. longibarbis and (b) D. urostriatum consist of 13 PCGs, 22 tRNA genes, and two rRNA genes. Genes encoded on the reverse strand are highlighted inside the circle, while those on the forward strand are marked outside the circle.

Table 1.
Detailed information on the D. longibarbis and D. urostraitum mitogenomes including the start and end positions of each gene, the number of overlapping and atp6-cox3 intergenic nucleotides, the strand positions of genes, as well as the start and stop codons of entire PCGs and the anticodons of tRNAs.
The overall base compositions of the complete mitogenomes of D. longibarbis and D. urostriatum are provided in Table 2. The AT content of the mitogenome is 59.32% in D. longibarbis and 58.73% in D. urostriatum, with respective values of 59.56% and 58.84% for PCGs, 56.74% and 57.03% for tRNAs, 57.01% and 56.61% for rRNAs, and 66.51% and 64.70% for the control region (Table 2).
Table 2.
Detailed information on the nucleotide composition of the whole mitogenome (Total) and its PCGs, tRNAs, rRNAs, and control region (C.R.) in D. longibarbis and D. urostriatum.
There were 13 intergenic sequences between adjacent genes within the mitogenomes of the two species, ranging in length from 1 to 30 bp, and seven overlap sequences, ranging in length from 1 to 13 bp (Table 1). The intergenic sequence between the atp6 and cox3 genes is a synapomorphy feature to Callichthyidae, with a length of 29 bp (TATTTAAATCTAGCTCTATTAAATTAATT) in D. longibarbis and 30 bp (TATCTAAAACTATACTAAACTAAATAATTA) in D. urostriatum (Table 1).
3.2. Protein-Coding Genes
In both D. longibarbis and D. urostriatum, the nad6 gene is located on the light (L) strand, and the 12 PCGs (atp6, atp8, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4l, nad5, and cytb) are located on the heavy (H) strand (Table 1). All 13 PCGs initiate with the conventional start codon ATG, except for the cox1 gene, which begins with GTG as its start codon. Six PCGs (nad1, nad2, atp6, atp8, nad4l, nad5, and nad6) terminate with complete stop codons (TAA or TAG), while the cox1 gene terminates with AGG (Table 1). In addition, cox2, nad4, and cytb terminate with an incomplete stop codon (T-), and cox3 terminates with an incomplete stop codon (TA-).
The AT skew of the 13 PCGs for D. longibarbis was zero or positive for nad2 (0.123), cox2 (0.079), atp8 (0.130), atp6 (0.040), cox3 (0.005), nad4 (0.000), and nad5 (0.048). For D. urostriatum, the AT skew was positive for nad2 (0.122), cox2 (0.091), atp8 (0.091), atp6 (0.014), nad4 (0.048), and nad5 (0.062), and negative for the other genes (Figure 2a,b). On the other hand, the GC skew was negative for both species, except for the nad6 gene (Figure 2a,b).
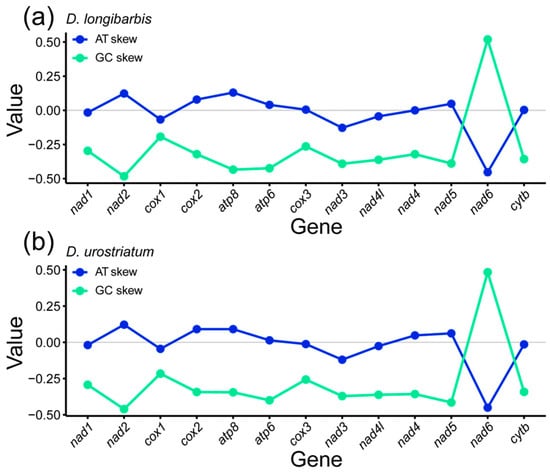
Figure 2.
AT and GC skewness of 13 PCGs in the mitogenomes of (a) D. longibarbis and (b) D. urostriatum.
Excluding the termination codon, the total number of codons in the 13 PCGs of both species was the same at 3795. For D. longibarbis, Leucine (Leu, 634), Alanine (Ala, 328), Isoleucine (Ile, 306), and Threonine (Thr, 303) were the most frequent, followed by Cysteine (Cys, 24), Arginine (Arg, 76), Aspartic Acid (Asp, 77), and Lysine (Lys, 84), which were the least frequent (Figure 3a and Table S3). In the case of D. urostriatum, Leucine (Leu, 635), Alanine (Ala, 321), Threonine (Thr, 314), and Isoleucine (Ile, 302) were the most frequent, and Cysteine (Cys, 24) Arginine (Arg, 76) Aspartic Acid (Asp, 78), and Lysine (Lys, 84) were the least frequent (Figure 3b and Table S4). The codon frequencies in the two species were similar, but the third most common codon was Ile in D. longibarbis and Thr in D. urostriatum.
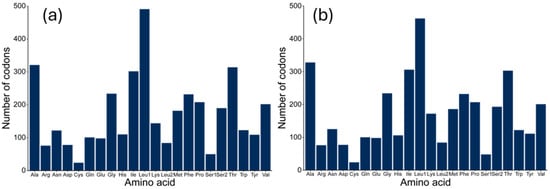
Figure 3.
Frequency of amino acids in the 13 PCGs of the (a) D. longibarbis and (b) D. urostriatum mitogenomes.
Regarding the RSCU results for D. longibarbis and D. urostriatum (Figure 4a,b and Tables S3 and S4), in D. longibarbis, CGA (Arg, 2.42), CUA (Leu1, 2.24), UCA (Ser2, 2.19), and CCA (Pro, 2.18) had the highest RSCU values, while in D. urostriatum, CUA (Leu1, 2.63), UCA (Ser2, 2.28), CCA (Pro, 2.08), and CGA (Arg, 2.00) had the highest RSCU values. On the other hand, ACG (Thr, 0.04), GCG (Ala, 0.05), UGG (Trp, 0.11), and CAG (Gln, 0.14) had the lowest RSCU values in D. longibarbis, followed by ACG (Thr, 0.08), GCG (Ala, 0.10), AAG (Lys, 0.12), and CCG (Pro, 0.12) in D. urostriatum (Figure 4a,b and Tables S3 and S4). In both species, a strong anti-bias G was analyzed at the third position of codons.
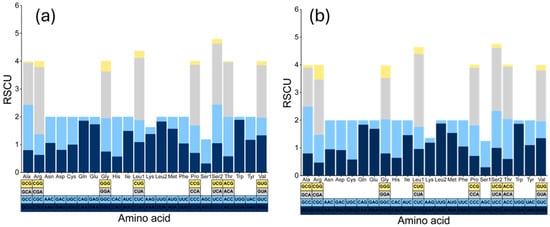
Figure 4.
Relative synonymous codon usage (RSCU) in the 13 PCGs of the (a) D. longibarbis and (b) D. urostriatum mitogenomes.
To identify hidden evolutionary information about D. longibarbis and D. urostriatum, genetic distance, nucleotide diversity, and Ka/Ks ratios were analyzed for their 13 PCGs among species belonging to the Callichthyidae (Figure 5 and Figure 6). In the nucleotide diversity (Pi) observed in this study, cox2 (0.100), cox3 (0.116), and cox1 (0.127) showed the lowest values, while nad3 (0.150), nad2 (0.150), and atp6 (0.149) showed the highest values (Figure 5). For K2P distance, cox2 (0.110), cox3 (0.131), and cox1 (0.143) had the lowest values, followed by nad3 (0.175), atp6 (0.173), and atp8 (0.173) (Figure 6). As a common result of Pi and K2P distance, the cox genes were found to have the highest conservation as it exhibited low genetic diversity and low intergenic distance. In addition, the genes with the lowest Ka/Ks ratio were cox1 (0.011), nad4l (0.018), and cox2 (0.021), and the genes with the highest Ka/Ks ratio were atp6 (0.115), nad2 (0.083), and nad6 (0.064) (Figure 6).
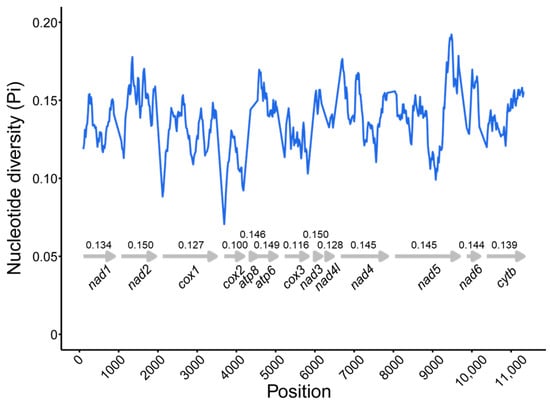
Figure 5.
The nucleotide diversity by position of concatenated 13 PCGs for 24 species in the Callichthyidae. The arrows indicate the position of each PCG, and the numbers indicate the pi value of that PCG. Detailed information about the species is provided in Table S1.
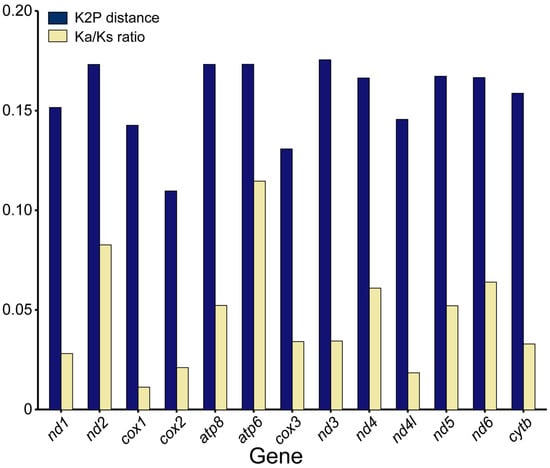
Figure 6.
K2P distance and Ka/Ks ratio for 13 PCGs belonging to 24 species of the Callichthyidae. Detailed information about the species is provided in Table S1.
3.3. tRNAs, rRNAs and Control Region
In both species, the lengths of 22 tRNAs ranged from 67 to 75 bp. Fourteen tRNAs (trnF, trnV, trnI, trnM, trnN, trnA, trnY, trnD, trnK, trnH, trnS1, trnL1, trnE, and trnT) were located on the H strand, while the remaining eight tRNAs (trnL2, trnQ, trnW, trnC, trnS2, trnG, trnR, and trnP) were located on the L strand (Table 1). The predicted secondary structure of tRNAs had a cloverleaf structure except for trnS1, which does not have dihydrouridine (DHU) arm structure (Figures S3 and S4).
For D. longibarbis, the rrnS was 949 bp long and the rrnL was 1641 bp, while for D. urostriatum, the rrnS was 950 bp and the rrnL was 1642 bp (Table 1). All rRNAs were located on the H strand.
Interestingly, the control regions of D. longibarbis and D. urostriatum were 869 and 864 bp in length, respectively, which is the smallest length observed, considering that the control regions of other Callichthyidae species range from 904 to 1299 bp in length (Figure S5).
3.4. Phylogenetic Analysis
The phylogenetic results were constructed using either Bayesian interference or the maximum likelihood estimate model, with 13 PCGs and 13 PCGs + 2rRNAs, incorporating species from three families within the suborder Loricarioidei (Callichthyidae, Loricariidae, and Trichomycteridae), and species from the family Siluridae within the suborder Siluroidei as outgroups (Figure 7, Figures S6 and S7). The phylogenetic tree using 13 PCGs and the tree using 13 PCGs + 2rRNA shared the same topology). Both the ML and BI trees constructed monophyletic clades for Callichthyidae, Trichomycteridae, Loricariidae, and Silluridae. Callichthyinae and Corydoradinae were separated within Callichthyidae with high posterior probability (BI = 1.0) and bootstrap percentage (ML = 100), forming a single clade. D. longibarbis and D. urostriatum were sister species to each other, clustered in the same clade with Hoplosternum littorale within Callichthyinae and had high support values (ML = 100 and BI = 1.0).
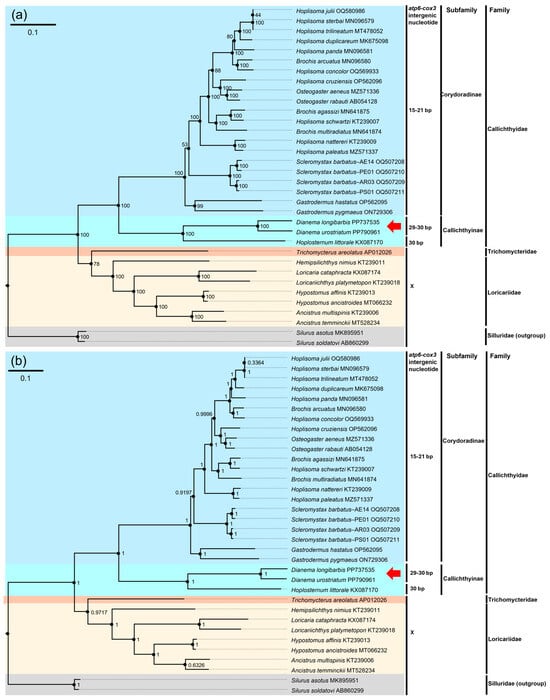
Figure 7.
The phylogenetic trees of 34 published Siluriformes mitogenomes, including that of D. longibarbis and D. urostriatum, constructed based on the concatenated nucleotide sequences of 13 PCGs and two rRNAs. (a) The numbers on the nodes indicate ML bootstrap percentages. (b) The numbers on the nodes indicate Bayesian posterior probability. GenBank accession number for the published sequence of each species is appended. The base pair numbers indicated the number of intergenic nucleotides between two genes, atp6 and cox3. The red arrow represents the two catfish species analyzed in this study. References for the mitogenome data used in this analysis are appended in Table S1.
4. Discussion
The mitogenomes of D. longibarbis and D. urostriatum, two species belonging to the Dianema observed in this study, were 16,493 and 16,495 bp in length, respectively, and consisted of 13 PCGs, two rRNAs, 22 tRNAs, and one control region. Their structure and arrangement were identical to those of common vertebrate mitogenomes [12]. In 13 PCGs, D. longibarbis and D. urostriatum contain the same initiation and termination codons, and additionally, the incomplete termination codons observed in cox2, cox3, nad4, and cytb are commonly observed in PCGs of teleost mitogenomes, and these incomplete termination codons are considered to function normally during post-transcriptional polyadenylation [36,37].
Insertion sequences of 29 bp (TATTTAAATCTAGCTCTATTAAATTAATT) and 30 bp (TATCTAAAACTATACTAAACTAAATAATTA) were observed in D. longibarbis and D. urostriatum, respectively. A similarly long intergenic sequence of 30 bp (CACATAATTAATCACATAAATTAAATCT) was also observed in H. littiorale, another species in the Callichthyinae, while a shorter intergenic sequence of 15–21 bp was observed in Corydoradinae [14,38,39,40,41,42,43,44,45,46,47,48]. Given the high interspecific similarity in Corydoradinae, it was suggested that the atp6-cox3 intergenic sequence could function as a molecular marker for species classification, whereas in Callichthyinae, no intergenic sequence similarity was observed, except for length [48]. The presence of long intergenic sequences in Callichthyinae could be due to sequence duplication, as previously hypothesized, but the lack of sequence identity between the genomes of Dianema and Hoplosternum suggests that duplication may not be the underlying cause [8]. Nevertheless, the reason for the longer intergenic sequences observed in Callichthyinae remains unclear, and it is also unknown where these intergenic sequences originated. Further studies of the mitogenomes of Callichthyinae species are needed to address the mystery of the long intergenic sequences in this subfamily.
Ka/Ks ratios below 1 were observed for all PCGs, indicating that the genes are under purifying selection [49]. Although in the previous comparison of PCGs belonging to the Corydoradinae, atp8 showed higher Pi and K2P distance values than cox1, the results of the analysis of PCGs in the Callichthyinae indicate that the atp8 gene in the Hoplosternum or Dianema genera is under high evolutionary pressure [38,45,48].
Deletion of the DHU arm in trnS1 is a common feature observed in a variety of teleosts, as well as previous studies of the Callichthyidae mitogenomes [36,38]. Despite being a non-coding region, the control region is known to contain promoter regions and is where the origin of replication is located [50]. Although all species in the Callichthyidae, except D. longibarbis and D. urostriatum, were found to have tandem repeat sequences in the upstream position of the control region, the absence of tandem repeats in the control region of these two species may account for the unusually short length of the control region in D. longibarbis and D. urostriatum.
Discordance among species in the genera of Brochis, Hoplisoma, and Osteogaster within the Corydoradinae was consistent with previous studies [38,45,48]. To date, studies of the evolutionary position of the Callichthyinae have not shown consistent results. For example, the cladogram based on osteological features recovered the topology of ((((Hoplosternum + Dianema) + Megalechis) + Leptophosternum) + Callichthys) [7], and the phylogenetic trees based on mitochondrial genes (two rRNAs, two tRNAs, and nad4) and mitochondrial genes (two rRNAs, two tRNAs, and nad4) + the nuclear gene (sia) followed the topology of ((((Megalechis + Leptophosternum) + Callichthys) + Hoplosternum) + Dianema) [8,9]. In addition, a cladogram constructed from morphological features and mitochondrial genes (two rRNAs, nad4, and cytb) + the nuclear gene (rag1) represented the topology of (((Hoplosternum + Dianema) + (Callichthys + (Megalechis + Leptophosternum)))) [15]. Phylogenetic classifications constructed using partial mitogenomes and partial mitogenomes + nuclear genes commonly indicate that Dianema is the primitive species of Callichthyinae. However, this study suggests that H. littorale is a primitive species within Callichthyinae. This discrepancy may result from the fact that only D. longibarbis, D. urostriatum, and H. littorale were included in this study, and further studies of the mitogenomes of Callichthyinae species are needed to resolve this issue.
5. Conclusions
This study describes the complete mitogenomes of D. longibarbis and D. urostriatum, the only species in the genus Dianema. The characteristic short length of the control region in Dianema is attributed to the absence of tandem repeats, which are common in other Callichthyidae species. Furthermore, the increased nucleotide diversity and divergence of the atp8 gene, compared to species in the Corydoradinae, suggest that the atp8 gene in the Callichthyinae is under strong evolutionary pressure. The constructed phylogenetic tree indicated that Callichthyinae and Dianema were supported as monophyletic groups based on strong support from both Bayesian inference (BI) and maximum likelihood (ML) analyses. Nevertheless, unlike previous studies, whether Dianema represents a primitive genus in the Callichthyidae requires further investigation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes16030355/s1, Figure S1: Photographs of (a) D. longibarbis and (b) D. urostriatum used for DNA extraction; Figure S2: The coverage depth for each genomic position in (a) D. longbibarbis and (b) D. urostriatum; Figure S3: The predicted secondary structure of tRNAs in the D. longibarbis mitogenome; Figure S4: The predicted secondary structure of tRNAs in the D. urostriatum mitogenome; Figure S5: Tandem repeat sequences in the control regions of 24 species in the Callichthyidae; Figure S6: A maximum likelihood (ML) phylogenetic tree of 34 published mitogenomes of Siluriformes, including mitogenomes of D. longibarbis and D. urostriatum, based on the concatenated nucleotide sequences of 13 PCGs; Figure S7: A Bayesian phylogenetic tree of 32 published mitogenomes of Siluriformes, including mitogenomes of D. longibarbis and D. urostriatum, based on the concatenated nucleotide sequences of 13 PCGs; Table S1: The mitogenome data used in phylogenetic analysis; Table S2: Best substitution model used for phylogenetic trees; Table S3: Details on codon count and RSCU in the D. longibarbis mitogenome; Table S4: Details on codon count and RSCU in the D. urostriatum mitogenome. References [14,38,40,41,42,43,44,45,46,47,48,51,52,53,54,55,56,57,58,59] are cited in the Supplementary Materials.
Author Contributions
S.D.D.: conceptualization, methodology, software, visualization, and writing; J.-S.R.: supervision, funding acquisition, reviewing, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Incheon National University Grant (2022-0101).
Institutional Review Board Statement
All animal study protocols were approved by the Institutional Review Board of Incheon National University (INU-ANIM-2024-1).
Informed Consent Statement
Not applicable.
Data Availability Statement
The Bioproject, Biosample, and SRA accession numbers for D. longibarbis are PRJNA1229777, SAMN47142742, and SRR32520581, respectively, while those for D. urostriatum are PRJNA1229778, SAMN47142749, and SRR32520608, respectively. The data supporting these findings are publicly available at the National Center for Biotechnology Information under the accession numbers PP737535 (D. longibarbis) and PP790961 (D. urostriatum).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Teugels, G.G. Taxonomy, phylogeny and biogeography of catfishes (Ostariophysi, Siluroidei): An overview. Aquat. Living Resour. 1996, 9, 9–34. [Google Scholar] [CrossRef]
- Armbruster, W.J. Global catfish biodiversity. Am. Fish. Soc. Symp. 2011, 77, 15–37. Available online: https://webhome.auburn.edu/~armbrjw/Global_Catfish.pdf (accessed on 28 February 2025).
- Nelson, J.; Grande, T.; Wilson, M. Fishes of the World, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; ISBN 9871118342336. [Google Scholar]
- Alexander, R. McN. Structure and function in the catfish. J. Zool. 1965, 148, 88–152. [Google Scholar] [CrossRef]
- Gee, J.H.; Graham, J.B. Respiratory and hydrostatic functions of the intestine of the catfishes Hoplosternum thoracatum and Brochis splendens (Callichthyidae). J. Exp. Biol. 1978, 74, 1–16. [Google Scholar] [CrossRef]
- Reis, R.E. Check List of the Freshwater Fishes of South and Central America; EDIPUCRS: Porto Alegre, Brazil, 2003; ISBN 85-7430-361-5. [Google Scholar]
- Reis, R.E. Anatomy and phylogenetic analysis of the neotropical Callichthyid catfishes (Ostariophysi, Siluriformes). Zool. J. Linn. Soc. 1998, 124, 105–168. [Google Scholar] [CrossRef]
- Shimabukuro-Dias, C.K.; Oliveira, C.; Reis, R.E.; Foresti, F. Molecular phylogeny of the armored catfish family Callichthyidae (Ostariophysi, Siluriformes). Mol. Phylogenet. Evol. 2004, 32, 152–163. [Google Scholar] [CrossRef]
- Mariguela, T.C.; Alexandrou, M.A.; Foresti, F.; Oliveira, C. Historical biogeography and cryptic diversity in the Callichthyinae (Siluriformes, Callichthyidae). J. Zool. Syst. Evol. Res. 2013, 51, 308–315. [Google Scholar] [CrossRef]
- Ruiz-Tafur, M.; Sánchez Riveiro, H.; García-Ayala, J. First record of Dianema urostriatum (Miranda Ribeiro, 1912) (Siluriformes: Callychthidae), in the Putumayo River, Amazon Basin, Peru. Folia Amaz. 2022, 31, 273–278. [Google Scholar] [CrossRef]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. Ecol. Evol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic. Acids. Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends. Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Parente, T.E.; Moreira, D.A.; Buckup, P.A.; de Andrade, P.C.C.; Magalhães, M.G.P.; Furtado, C.; Britto, M.R.; Val, A.L. Remarkable genetic homogeneity supports a single widespread species of Hoplosternum littorale (Siluriformes, Callichthyidae) in South America. Conserv. Genet. Resour. 2018, 10, 563–569. [Google Scholar] [CrossRef]
- Vera-Alcaraz, H.S. Relações Filogenéticas das Espécies da Família Callichthyidae (Ostariophysi, Siluriformes). Ph.D. Thesis, Pontifical Catholic University of Rio Grande do Sul, Porto Alegre, Brazil, 2013. Available online: https://tede2.pucrs.br/tede2/handle/tede/263 (accessed on 17 March 2025).
- Nam, S.-E.; Kim, J.; Rhee, J.-S. First complete mitochondrial genome from family Moinidae, Moina macrocopa (Straus, 1820) (Cladocera; Moinidae). Mitochondrial DNA B Resour. 2022, 7, 980–982. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 17 January 2025).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic. Acids. Res. 2017, 45, e18. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.Y.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic. Acids. Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. TRNAscan-SE On-Line: Integrating search and context for analysis of transfer RNA genes. Nucleic. Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree. Tree Figure Drawing Tool. 2009. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 2 February 2025).
- Satoh, T.P.; Miya, M.; Mabuchi, K.; Nishida, M. Structure and variation of the mitochondrial genome of fishes. BMC Genom. 2016, 17, 719. [Google Scholar] [CrossRef]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Translational readthrough potential of natural termination codons in eucaryotes—The impact of RNA sequence. RNA Biol. 2015, 12, 950–958. [Google Scholar] [CrossRef]
- Sun, C.H.; Huang, Q.; Zeng, X.S.; Li, S.; Zhang, X.L.; Zhang, Y.N.; Liao, J.; Lu, C.H.; Han, B.P.; Zhang, Q. Comparative analysis of the mitogenomes of two Corydoras (Siluriformes, Loricarioidei) with nine known corydoras, and a phylogenetic analysis of Loricarioidei. Zookeys 2022, 2022, 89–107. [Google Scholar] [CrossRef]
- Lv, L.; Su, H.; Xu, B.; Liu, Q.; Xiao, T. Complete mitochondrail genome of Corydoras agassizii. Mitochondrial DNA Part B Resour. 2020, 5, 727–728. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, B.; Xiao, T.; Liu, Q. Characterization and phylogenetic analysis of Corydoras arcuatus mitochondrial genome. Mitochondrial DNA B Resour. 2019, 4, 2876–2877. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, B.; Xiao, T. Complete mitochondrial genome of Corydoras duplicareus (Teleostei, Siluriformes, Callichthyidae). Mitochondrial DNA B Resour. 2019, 4, 1832–1833. [Google Scholar] [CrossRef]
- Chen, L.; Xu, B.; Xiao, T.; Liu, Q. Characterization and phylogenetic analysis of Corydoras trilineatus mitochondrial genome. Mitochondrial DNA B Resour. 2020, 5, 3017–3018. [Google Scholar] [CrossRef]
- Moreira, D.A.; Buckup, P.A.; Britto, M.R.; Magalhães, M.G.P.; De Andrade, P.C.C.; Furtado, C.; Parente, T.E. The complete mitochondrial genome of Corydoras nattereri (Callichthyidae: Corydoradinae). Neotrop. Ichthyol. 2016, 14, e150167. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Y.; Xiao, T.; Xu, B. Complete mitochondrail genome of Corydoras panda (Teleostei, Siluriformes, Callichthyidae, Corydoradinae). Mitochondrial DNA B Resour. 2019, 4, 2878–2879. [Google Scholar] [CrossRef]
- Qiao, Z.; Liu, S.; Wang, S.; Li, T.; Han, Y. Complete mitochondrial genomes of two Corydoras (Siluriformes, Callichthyidae) and their phylogenetic implications. Pakistan J. Zool. 2024, 1–9. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, L.A.; Zhang, W. First complete mitochondrial genome of the Corydoras pygmaeus (Actinopteri: Callichthyidae) and its phylogenetic implications. Mitochondrial DNA B Resour. 2022, 7, 1688–1690. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Y.; Xu, B.; Xiao, T. Next-generation sequencing yields the complete mitochondrial genome of Corydoras sterbai (Teleostei, Siluriformes, Callichthyidae, Corydoradinae). Mitochondrial DNA B Resour. 2019, 4, 2880–2881. [Google Scholar] [CrossRef]
- Do, S.D.; Rhee, J.-S. First description of intergenic sequences in corydoradinae and introducing the complete mitogenome of Hoplisoma concolor (Siluriformes: Callichthyidae). Genes 2025, 16, 282. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinf. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Bronstein, O.; Kroh, A.; Haring, E. Mind the Gap! The mitochondrial control region and its power as a phylogenetic marker in Echinoids. BMC Evol. Biol. 2018, 18, 80. [Google Scholar] [CrossRef]
- Xu, B.; Su, H.; Liu, Q.; Lv, L.; Chen, K.; Xiao, T. Complete mitochondrial genome of Brochis multiradiatus. Mitochondrial DNA B Resour. 2020, 5, 646–647. [Google Scholar] [CrossRef]
- Moreira, D.A.; Buckup, P.A.; Furtado, C.; Val, A.L.; Schama, R.; Parente, T.E. Reducing the information gap on Loricarioidei (Siluriformes) mitochondrial genomics. BMC Genom. 2017, 18, 345. [Google Scholar] [CrossRef]
- Dalcin, R.H.; De La Ossa-Guerra, L.E.; Artoni, R.F.; Abilhoa, V. Complete mitochondrial genome of four Scleromystax barbatus (Siluriformes: Callichthyidae) populations. Neotrop. Ichthyol. 2023, 21, 1–11. [Google Scholar] [CrossRef]
- Saitoh, K.; Miya, M.; Inoue, J.G.; Ishiguro, N.B.; Nishida, M. Mitochondrial genomics of Ostariophysan fishes: Perspectives on phylogeny and biogeography. J. Mol. Evol. 2003, 56, 464–472. [Google Scholar] [CrossRef]
- Nakatani, M.; Miya, M.; Mabuchi, K.; Saitoh, K.; Nishida, M. Evolutionary history of Otophysi (Teleostei), a major clade of the modern freshwater fishes: Pangaean origin and Mesozoic radiation. BMC Evol. Biol. 2011, 11, 177. [Google Scholar] [CrossRef]
- Meng, F.; Yin, X.; Zhang, T.; Zhao, C.; Xue, X.; Xia, X.; Zhu, X.; Duan, Z.; Liu, B.; Liu, Y. The first determination and analysis of the complete mitochondrial genome of Ancistrus temmincki (Siluriformes: Loricariidae). Mitochondrial DNA B Resour. 2021, 6, 1583–1585. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Reis, D.A.; Pasa, R.; Menegidio, F.B.; Heslop-Harrison, J.S.; Schwarzacher, T.; Kavalco, K.F. The complete mitochondrial genome of two armored catfish populations of the genus Hypostomus (Siluriformes, Loricariidae, Hypostominae). Front. Ecol. Evol. 2020, 8, 4–9. [Google Scholar] [CrossRef]
- Yang, N.; Li, Y.; Liu, Z.; Chen, Q.; Shen, Y. The complete mitochondrial genome of Silurus asotus (Siluriformes: Siluridae: Silurus) and its phylogenetic analysis. Mitochondrial DNA B Resour. 2019, 4, 2377–2378. [Google Scholar] [CrossRef]
- Wang, K.; Xu, J.; Cui, J.; Li, Q.; Xu, P.; Sun, X. Complete mitochondrial genome of Northern Sheatfish (Silurus soldatovi). Mitochondrial DNA 2015, 26, 891–892. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).