1. Introduction
The Chinese soft-shelled turtle (
P. sinensis) is an economically important freshwater aquaculture reptile in China [
1], widely distributed across East and Southeast Asia [
2]. According to the Fisheries Statistical Yearbook of China, the aquaculture production of
P. sinensis reached 370,000 tons in 2022. Currently, the main species of soft-shelled turtles farmed in China include the native
P. sinensis, introduced species, newly bred varieties, and hybrid varieties [
3]. The native
P. sinensis is a geographically differentiated subspecies that has gradually evolved through long-term geographical isolation across various regions of China. The introduced species refers to those brought from abroad and selectively bred over multiple generations [
4]. New varieties [
5] are developed by selecting individuals with beneficial mutations from the native
P. sinensis population and breeding them through several generations. Hybrid varieties are produced by crossing two populations with a significant genetic distance [
6]. With the rapid development of the
P. sinensis farming industry, the market has become increasingly mixed with native, introduced, newly bred, and hybrid varieties, resulting in genetic degradation and a decline in genetic traits, which severely threatens the sustainable development of the industry. Meanwhile, many farmed
P. sinensis organisms have escaped into natural waters, and combined with overfishing and the impact of environmental disasters, the population of wild
P. sinensis has dramatically decreased, leading to a severe loss of genetic resources. In 2000, the wild
P. sinensis was listed as an endangered species in the International Union for Conservation of Nature (IUCN) Red List of Threatened Species [
6]. Understanding the genetic diversity and structure of populations aids in regulating hybrid breeding, preventing overfishing, and conserving germplasm resources, thereby ensuring the sustainable exploitation of the population [
7]. However, there is currently limited research on the genetic differences among different populations of the
P. sinensis, and due to technical limitations, the findings from various studies are often contradictory [
8]. Therefore, to standardize the management of
P. sinensis farming populations and make rational use of their genetic resources, there is an urgent need to analyze the genetic diversity of the main farming populations of
P. sinensis.
Morphological methods are the most traditional and direct approach for analyzing population genetic variation and have been widely applied in studies on the variation and identification of
P. sinensis populations. However, since morphological traits are influenced by both genetic factors and external environmental conditions and can be affected by growth stages and data processing methods, they must be combined with genetic molecular markers to objectively evaluate the genetic diversity of populations [
9]. With the continuous advancement of high-throughput sequencing technology, molecular marker-based approaches for population genetic analysis and species identification have become increasingly mainstream [
10]. Among the various types of molecular markers, single nucleotide polymorphisms (SNPs) and simple sequence repeats (SSRs, also known as microsatellites) are considered the most promising. SNPs refer to polymorphisms in DNA sequences caused by single-base transitions or transversions, resulting in genetic variation. They are characterized by high genetic stability, widespread distribution across the genome, and a typical biallelic nature, making them highly suitable for genetic studies [
11]. On the other hand, SSRs are simple tandem repeat sequences uniformly distributed throughout eukaryotic genomes, consisting of 2–6 nucleotide repeat motifs. SSR markers are highly polymorphic, abundant, and reproducible, and they require only small amounts of DNA for analysis [
12]. Given these characteristics, researchers can select the appropriate molecular marker based on laboratory conditions and the specific requirements of their study. To date, SSR markers have been widely applied in the analysis of genetic variation and species identification in aquatic animals [
13,
14,
15]. In the evaluation of genetic diversity in
P. sinensis populations, molecular markers such as mitochondrial genes [
6], SSRs [
16,
17], and SNPs [
18] have been used. However, there has been no precedent for studies combining morphological analysis with these methods, and the number of populations studied has been limited, so conducting genetic differentiation analysis between populations based on both phenotypic and genotypic data provides a more comprehensive and reliable assessment of the genetic structure.
Therefore, to objectively evaluate the genetic diversity and genetic structure of P. sinensis farming populations and to standardize their management, this study employs both morphological analysis and microsatellite markers to assess the genetic diversity across six different populations and develop germplasm identification markers, providing a reference for scientific breeding of P. sinensis.
4. Discussion
Morphological analysis is one of the effective methods for studying genetic variation within species [
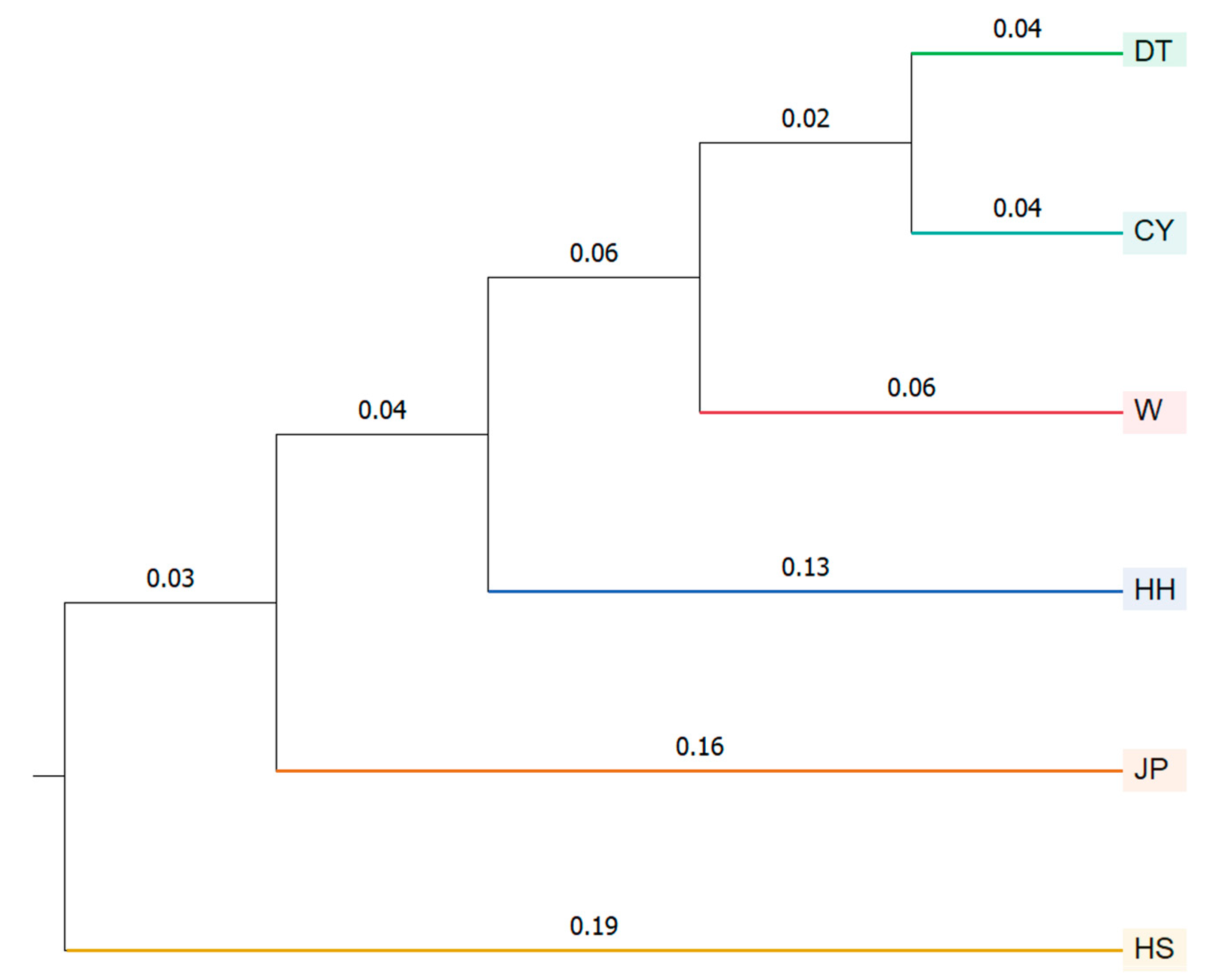
27]. In this study, cluster analysis based on morphological traits revealed that the JP and HH populations were grouped together; the DT and CY populations first clustered together and then with the W population; and, finally, the remaining populations clustered with the HS population. This result is consistent with the UPGMA tree analysis, where there is a close correlation between morphological development and genetic relationships. Discriminant analysis based on morphological traits was conducted for the six populations, and a discriminant function was established, resulting in an overall discriminant accuracy of 71.4%. The highest accuracy was observed in the DT and HH populations. Some individuals from the JP population were misclassified as belonging to the HH or HS populations, while a significant proportion of the CY and W populations were misclassified as DT and HH populations. The results of the cluster and discriminant analyses indicate a certain morphological similarity between the HH, JP, and HS populations, while the DT, CY, and W populations showed minor morphological differences. This morphological similarity also suggests a potential phylogenetic relationship between the populations [
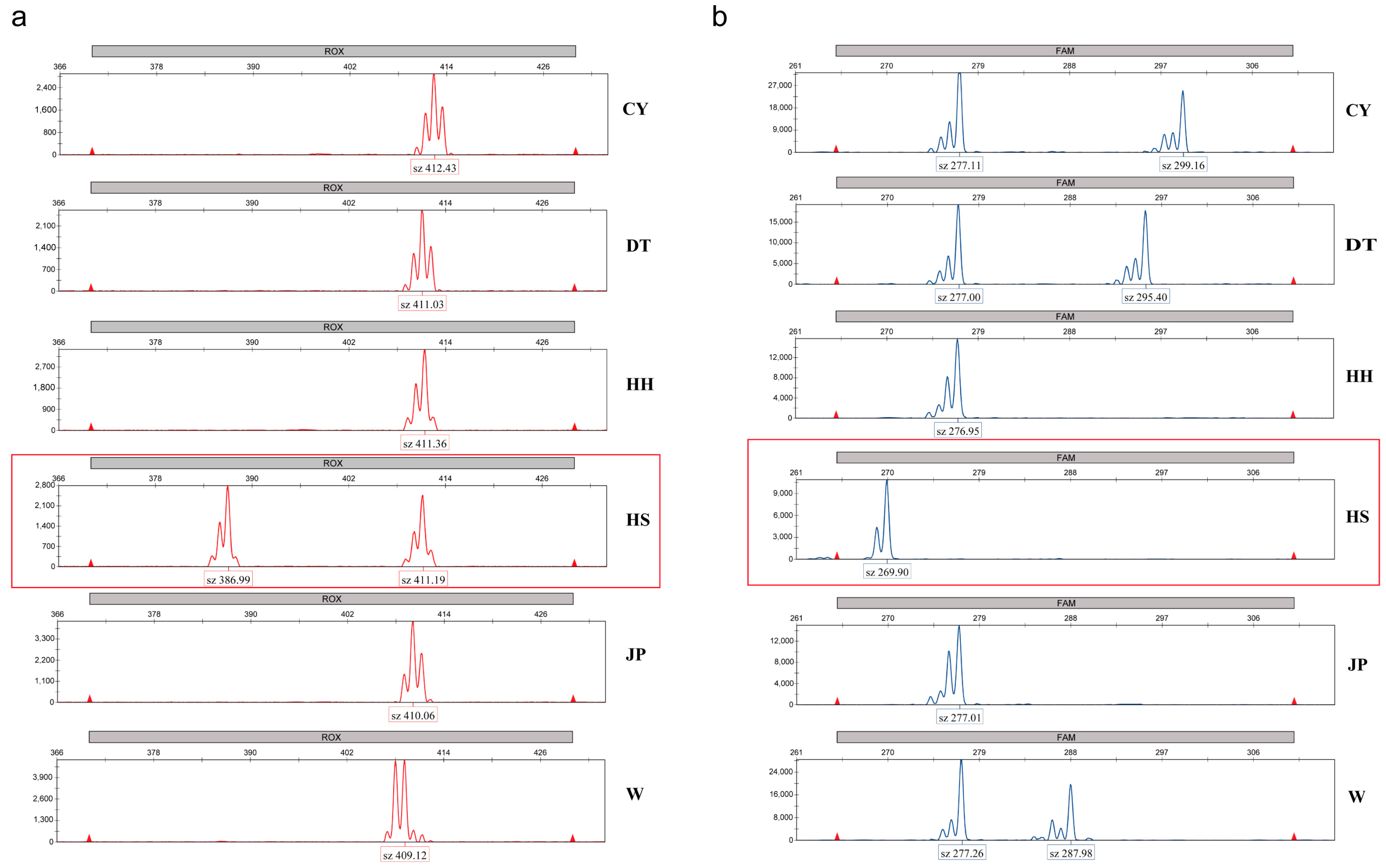
28]. Additionally, two microsatellite markers were identified in this study that can be used to distinguish the HS population, providing a reference for future germplasm identification in conjunction with the discriminant function.
Higher genetic diversity within a species increases its adaptability to environmental changes and enhances its evolutionary potential, making the study of genetic diversity a foundational aspect of breeding research [
29]. In this study, 12 highly polymorphic SSR markers with PIC values ranging from 0.033 to 0.891, He values between 0.032 and 0.801, and Na values ranging from 1.833 to 12.000 were developed based on Super-GBS sequencing to analyze the genetic diversity of six
P. sinensis populations. Compared to the 15 SSR markers (Na range: 2–7) developed by Que [
30], the SSR markers developed in this study exhibit higher polymorphism.
Genetic diversity parameters such as Na, Ne, Ho, and He reflect the genetic variation within and between populations or individuals. Among these, He is a key indicator of genetic diversity, as it is calculated based on allele frequencies and is less affected by sample size, providing a reliable measure of a population’s genetic diversity [
31]. In this study, the CY population exhibited the highest genetic diversity, while the HH population had the lowest. The genetic diversity of the JP population was higher than that of the HH population, which is consistent with the findings of Gang [
8]. In contrast, Zhang [
32] studied several
P. sinensis varieties using RAPD technology and concluded that the JP population had higher genetic diversity than the W population. However, this study reached the opposite conclusion, demonstrating that SSR markers provide greater stability and specificity compared to RAPD. The observed heterozygosity (Ho) ranged from 0.375 to 0.572, while the expected heterozygosity (He) was consistently higher than Ho. Additionally, the Fis values for all populations were greater than zero, indicating the presence of heterozygote deficiency. This phenomenon may be attributed to factors such as artificial selection, inbreeding, or small population sizes.
Genetic differentiation among six
P. sinensis populations was analyzed based on 12 microsatellite loci. The results of the AMOVA analysis revealed significant genetic variation among populations (
p < 0.05) and moderate gene flow (1 < Nm < 4). The majority of genetic variation was observed within individuals, whereas Zhang [
32] reported that genetic variation in
P. sinensis primarily occurs within populations. Moderate gene flow was generally detected among populations (1 ≤ Nm < 4), with particularly strong gene exchange among the DT, W, and CY populations (4 < Nm). Furthermore, the Hardy–Weinberg equilibrium analysis revealed significant deviations from equilibrium at multiple SSR loci across all populations. Comparatively, the HH population exhibited lower genetic diversity and relatively weaker gene exchange with the other populations. The primary habitat of the HH population is located in the Yellow River Basin [
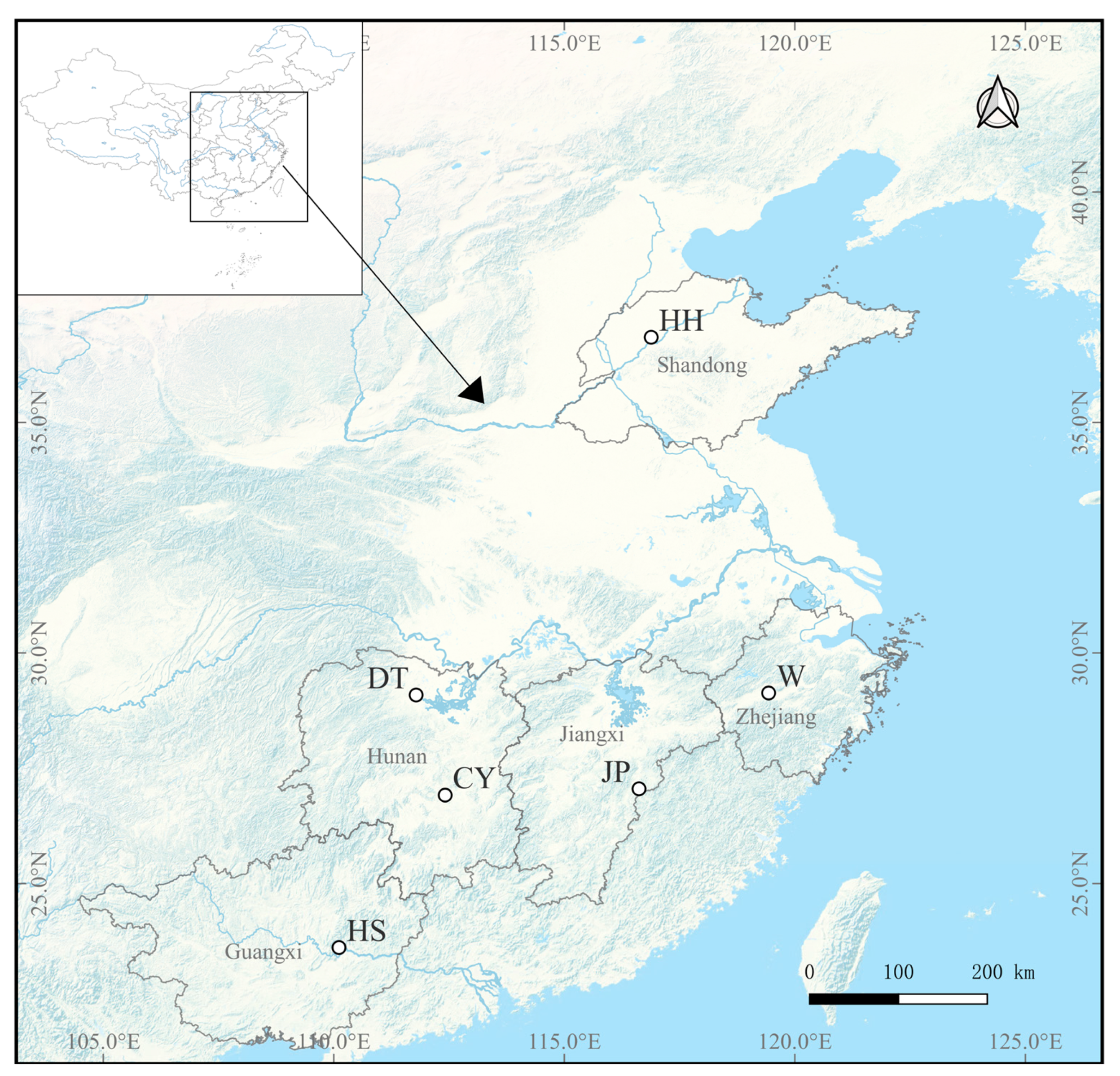
33], and its geographic isolation from the other populations likely restricts gene flow. The CY population, originally from Changyong Village in Hengyang County, Hunan Province, was formed by local villagers who spontaneously bred wild
P. sinensis. The W population was artificially selected from color-varied individuals of the DT population, forming a new variety [
17]. Therefore, both the CY and W populations have undergone a certain degree of artificial selection. The three populations exhibited highly frequent gene flow (
Nm > 4). While frequent gene exchange enhances genetic diversity, imbalanced migration rates and limited effective population sizes may lead to genetic drift, ultimately causing deviations from equilibrium. Fis is a key indicator for assessing genetic dynamics within a population. A positive Fis value (Fis > 0) suggests a deficiency of heterozygotes, whereas a negative Fis value (Fis < 0) indicates an excess of heterozygotes [
34]. In this study, the Fis values for all populations were greater than zero. Among the 12 SSR markers analyzed, 4 exhibited negative Fis values, while the remaining markers had positive values, indicating the presence of heterozygote deficiency in
P. sinensis populations. Despite the occurrence of frequent gene flow among populations, heterozygote deficiency persisted, which may be attributed to inbreeding, artificial selection pressure during aquaculture, or genetic drift resulting from small population sizes.
The F
ST is an important indicator for measuring the degree of genetic differentiation among populations or subpopulations, and it is negatively correlated with gene flow. According to Wright [
35], when F
ST ≥ 0.05, it indicates moderate genetic differentiation between populations. From the results in
Table 5, except for the F
ST between the CY, W, and DT populations, which is less than 0.05, all other population pairs showed F
ST values greater than 0.05. This suggests that, despite frequent gene flow, most populations still exhibit moderate genetic differentiation. AMOVA analysis also revealed significant genetic variation among populations. However, the excessively frequent gene exchange between the CY, W, and DT populations has suppressed genetic differentiation between these groups. The magnitude of genetic distance is an important factor in studying population genetic diversity and is positively correlated with population divergence time. Analysis of the genetic distances between populations showed that the HS population had the greatest genetic distance from the HH and JP populations, while the DT and CY populations exhibited the smallest genetic distance. Crossbreeding between populations with greater genetic distances is more likely to result in heterosis, which can provide valuable insights for future hybrid breeding studies [
36].
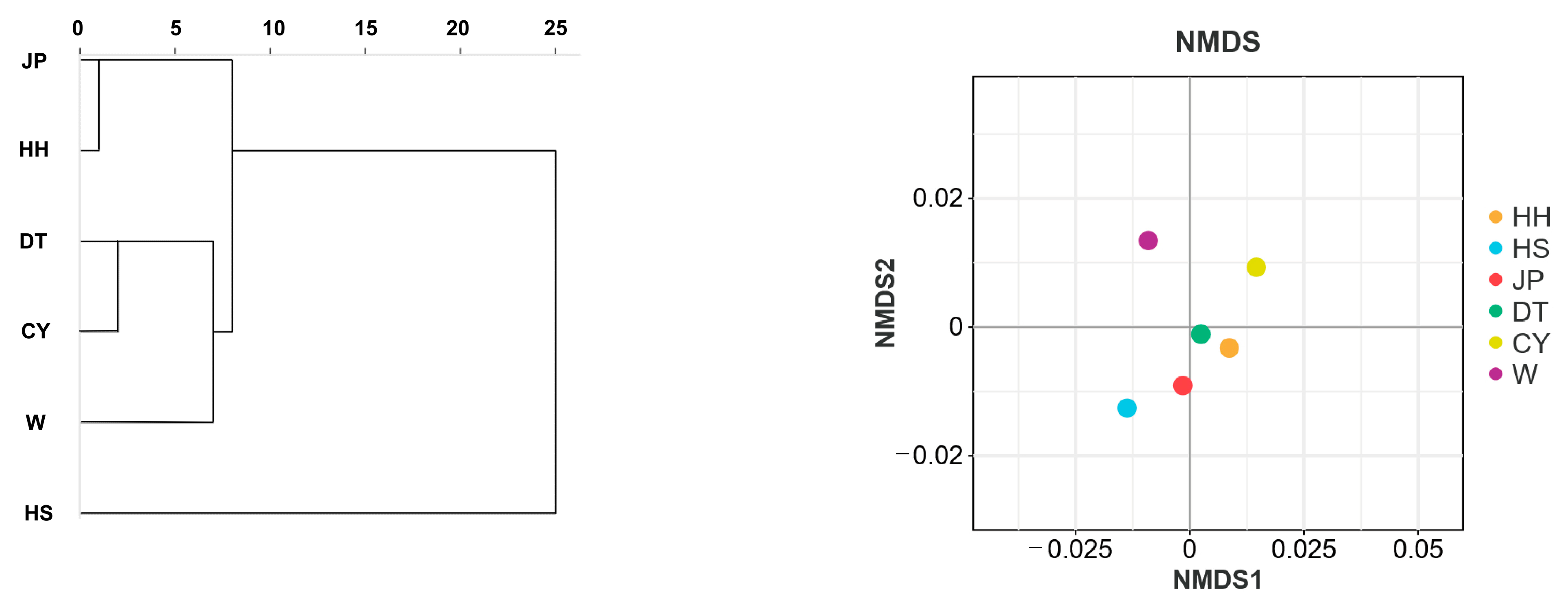
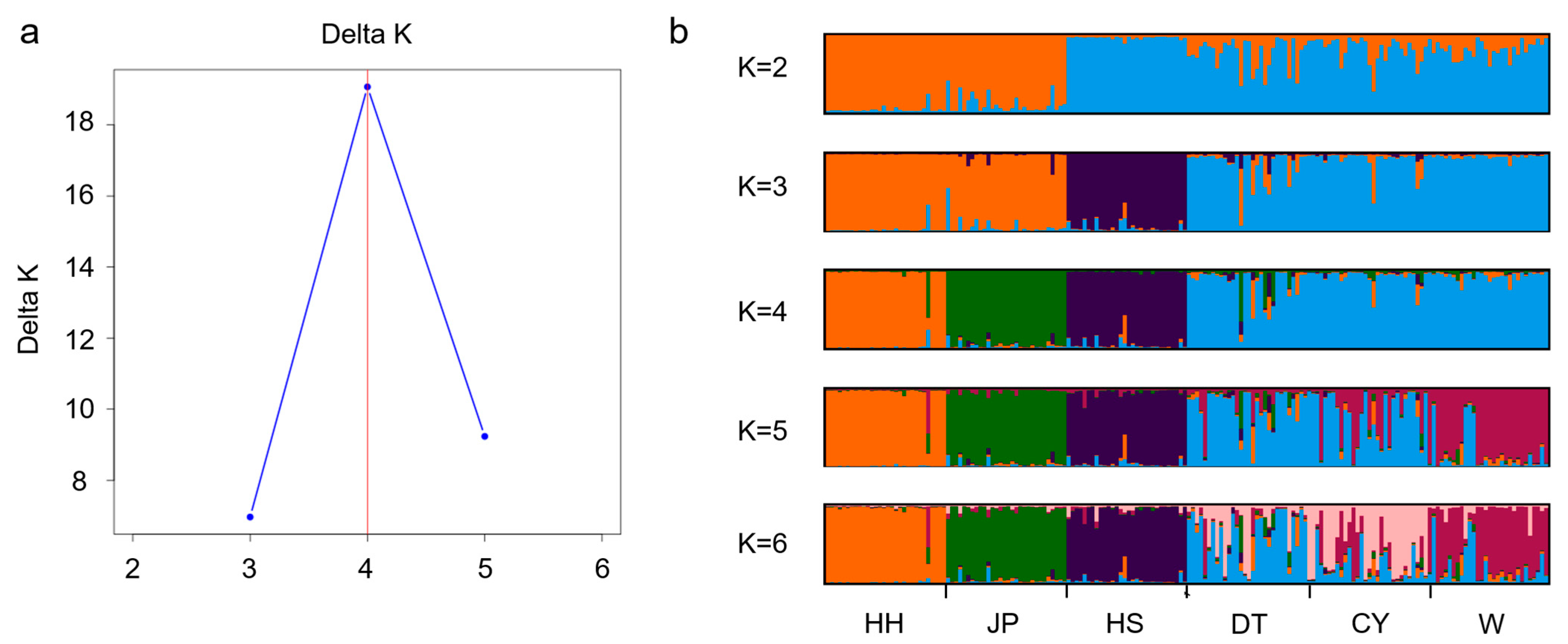
The STRUCTURE analysis divided the six
P. sinensis populations into four subgroups. In the phylogenetic tree analysis, the DT population clustered with the CY population first, followed by the W population, then the JP and HH populations, and finally the HS population. This clustering pattern is consistent with the morphological clustering analysis results. In the PCoA results, the HS population was consistently assigned to a separate cluster. Based on the integrated findings, the HS population was classified as one group; HH and JP as another; and DT, W, and CY as a third group. This classification aligns with the results of previous studies conducted in our laboratory and is consistent with similar conclusions drawn by Gang [
8]. The HH, DT, and HS populations represent the native
P. sinensis of China. The JP population, on the other hand, is a new variety that was developed after multiple generations of selective breeding from the native
P. sinensis imported from Japan [
17]. The fact that the HH and JP populations clustered together may be attributed to the Japanese population’s origin from the Chinese
P. sinensis, which was historically introduced as food to Japan, followed by long-term geographic isolation. The Yellow River
P. sinensis, native to the Yellow River Basin, is geographically close to Japan, and it is hypothesized that the Japanese population originated from the Yellow River
P. sinensis and underwent genetic differentiation due to long-term isolation. However, despite this geographic proximity, the two populations have undergone substantial genetic differentiation (0.15 < F
ST) [
33]. There has been no prior research specifically on the HS population, which originates from Guangxi. The
P. parviformis is a type of turtle from Quanzhou in Guangxi. Some studies classify the “small turtle” as a subspecies of
P. sinensis, while others consider it a subspecies without reproductive isolation from
P. sinensis [
37]. In this study, the HS population was distinguished from the other populations and categorized into a separate subgroup. Given that both the HS population and the
P. parviformis are distributed in the Guangxi region and are geographically close, it is plausible to hypothesize that there might be a phylogenetic relationship between the HS population and the
P. parviformis. This hypothesis, however, requires further investigation. Additionally, all samples collected in this study were from artificially cultured populations, thus introducing certain limitations to the research. Future studies should include a greater number of wild samples to enhance the comprehensiveness and robustness of the findings.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}