
Sustained Epigenetic Reactivation in Fragile X Neurons with an RNA-Binding Small Molecule

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Quantitative Real-Time Polymerase Chain Reaction
2.2. Western Blot
2.3. ViewRNA Cell Plus
2.4. Cell Culture and Neuronal Differentiation
2.5. CUT&RUN
2.6. RNA-seq
2.7. Dendritic Spine Imaging
2.8. BrdU Immunofluorescence
2.9. Microscopy
3. Results
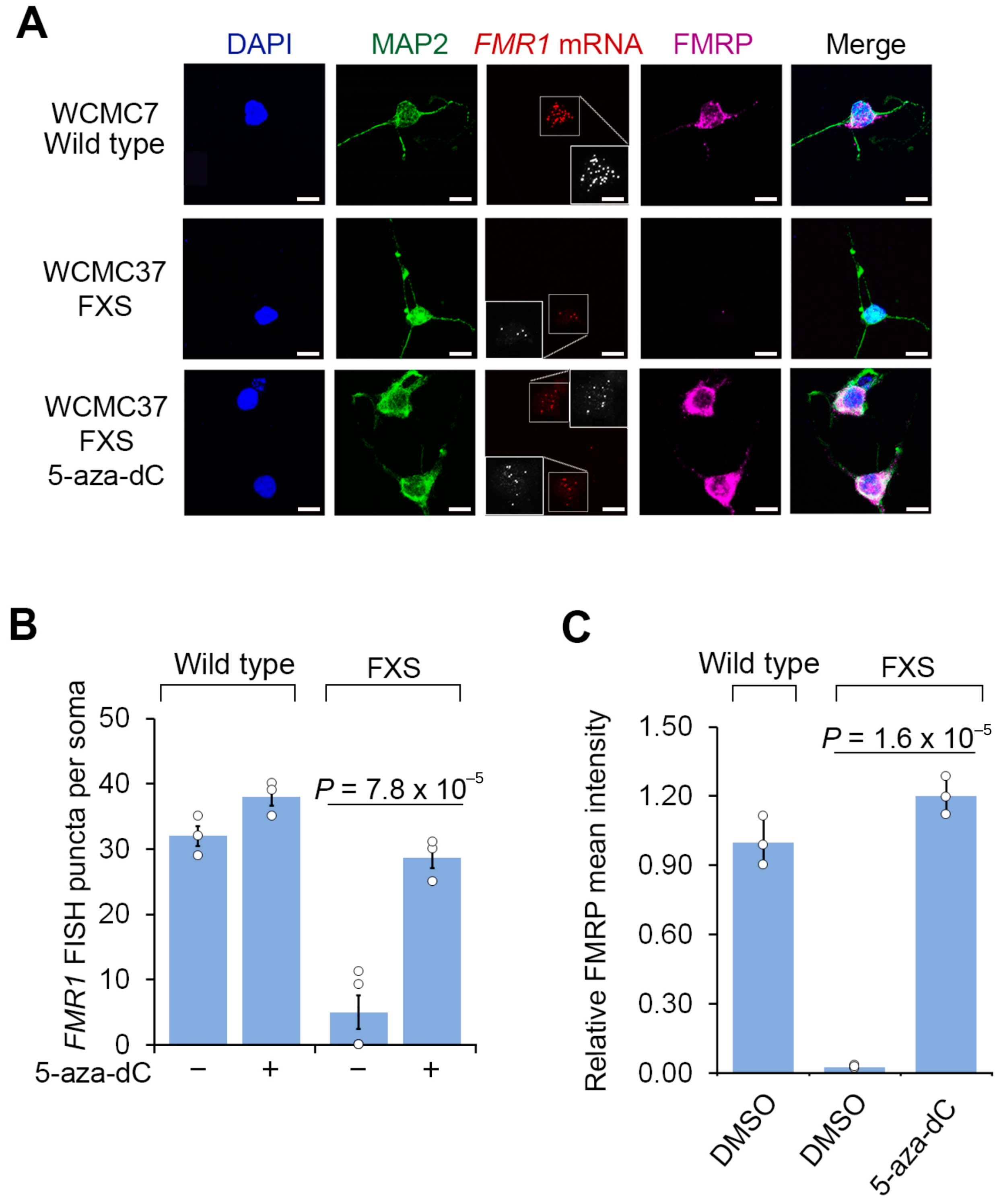
3.1. 5-aza-dC Restores FMRP Expression in Post-Mitotic Neurons
3.2. A CGG Repeat RNA-Binding Small Molecule Maintains FMR1 Reactivation in Neurons
3.3. 2HE-5NMe Selectively Maintains Epigenetic Activation of theFMR1 Locus
3.4. 2HE-5NMe Reverses Dendritic Spine Abnormalities in FXS Neurons
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oberlé, I.; Rousseau, F.; Heitz, D.; Kretz, C.; Devys, D.; Hanauer, A.; Boué, J.; Bertheas, M.F.; Mandel, J.L. Instability of a 550-Base Pair DNA Segment and Abnormal Methylation in Fragile X Syndrome. Science 1991, 252, 1097–1102. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Coffee, B.; Zhang, F.; Ceman, S.; Warren, S.T.; Reines, D. Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile x syndrome. Am. J. Hum. Genet. 2002, 71, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Bontekoe, C.J.M.; Severijnen, L.-A.; Oostra, B.A. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum. Genet. 2002, 110, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Irwin, S.A.; Patel, B.; Idupulapati, M.; Harris, J.B.; Crisostomo, R.A.; Larsen, B.P.; Kooy, F.; Willems, P.J.; Cras, P.; Kozlowski, P.B.; et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: A quantitative examination. Am. J. Med. Genet. 2001, 98, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Colak, D.; Zaninovic, N.; Cohen, M.S.; Rosenwaks, Z.; Yang, W.-Y.; Gerhardt, J.; Disney, M.D.; Jaffrey, S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 2014, 343, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, M.; Schuffenhauer, A.; Fruh, I.; Klein, J.; Thiemeyer, A.; Rigo, P.; Gomez-Mancilla, B.; Heidinger-Millot, V.; Bouwmeester, T.; Schopfer, U.; et al. High-Throughput Screening Using iPSC-Derived Neuronal Progenitors to Identify Compounds Counteracting Epigenetic Gene Silencing in Fragile X Syndrome. J. Biomol. Screen. 2015, 20, 1101–1111. [Google Scholar] [CrossRef]
- Kumari, D.; Swaroop, M.; Southall, N.; Huang, W.; Zheng, W.; Usdin, K. High-Throughput Screening to Identify Compounds That Increase Fragile X Mental Retardation Protein Expression in Neural Stem Cells Differentiated from Fragile X Syndrome Patient-Derived Induced Pluripotent Stem Cells. Stem Cells Transl. Med. 2015, 4, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Chiurazzi, P.; Pomponi, M.G.; Willemsen, R.; Oostra, B.A.; Neri, G. In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum. Mol. Genet. 1998, 7, 109–113. [Google Scholar] [CrossRef]
- Kumari, D.; Usdin, K. Polycomb group complexes are recruited to reactivated FMR1 alleles in Fragile X syndrome in response to FMR1 transcription. Hum. Mol. Genet. 2014, 23, 6575–6583. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Usdin, K. Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethylation of H3K27. Hum. Mol. Genet. 2016, 25, 3689–3698. [Google Scholar] [CrossRef]
- Li, M.; Zhao, H.; Ananiev, G.E.; Musser, M.T.; Ness, K.H.; Maglaque, D.L.; Saha, K.; Bhattacharyya, A.; Zhao, X. Establishment of Reporter Lines for Detecting Fragile X Mental Retardation (FMR1) Gene Reactivation in Human Neural Cells. Stem Cells Dayt. Ohio 2017, 35, 158–169. [Google Scholar] [CrossRef]
- Schermelleh, L.; Spada, F.; Easwaran, H.P.; Zolghadr, K.; Margot, J.B.; Cardoso, M.C.; Leonhardt, H. Trapped in action: Direct visualization of DNA methyltransferase activity in living cells. Nat. Methods 2005, 2, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, L.; Chen, P.; Xu, Z.; Qiu, J.; Ge, J.; Yu, K.; Zhuang, J. Brca1 Is Upregulated by 5-Aza-CdR and Promotes DNA Repair and Cell Survival, and Inhibits Neurite Outgrowth in Rat Retinal Neurons. Int. J. Mol. Sci. 2018, 19, 1214. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Sweatt, J.D. Covalent Modification of DNA Regulates Memory Formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef]
- Pan, Y.; Daito, T.; Sasaki, Y.; Chung, Y.H.; Xing, X.; Pondugula, S.; Swamidass, S.J.; Wang, T.; Kim, A.H.; Yano, H. Inhibition of DNA Methyltransferases Blocks Mutant Huntingtin-Induced Neurotoxicity. Sci. Rep. 2016, 6, 31022. [Google Scholar]
- Yang, W.-Y.; He, F.; Strack, R.L.; Oh, S.Y.; Frazer, M.; Jaffrey, S.R.; Todd, P.K.; Disney, M.D. Small Molecule Recognition and Tools to Study Modulation of r(CGG)(exp) in Fragile X-Associated Tremor Ataxia Syndrome. ACS Chem. Biol. 2016, 11, 2456–2465. [Google Scholar] [CrossRef] [PubMed]
- Bowler, E.H.; Bell, J.; Divecha, N.; Skipp, P.; Ewing, R.M. Proteomic Analysis of Azacitidine-Induced Degradation Profiles Identifies Multiple Chromatin and Epigenetic Regulators Including Uhrf1 and Dnmt1 as Sensitive to Azacitidine. J. Proteome Res. 2019, 18, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef]
- Nair, V.P.; Liu, H.; Ciceri, G.; Jungverdorben, J.; Frishman, G.; Tchieu, J.; Cederquist, G.Y.; Rothenaigner, I.; Schorpp, K.; Klepper, L.; et al. Activation of HERV-K(HML-2) disrupts cortical patterning and neuronal differentiation by increasing NTRK3. Cell Stem Cell 2021, 28, 1566–1581.e8. [Google Scholar] [CrossRef] [PubMed]
- Riessland, M.; Kolisnyk, B.; Kim, T.W.; Cheng, J.; Ni, J.; Pearson, J.A.; Park, E.J.; Dam, K.; Acehan, D.; Ramos-Espiritu, L.S.; et al. Loss of SATB1 Induces p21-Dependent Cellular Senescence in Post-mitotic Dopaminergic Neurons. Cell Stem Cell 2019, 25, 514–530.e8. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zhang, X.-J.; Renier, N.; Wu, Z.; Atkin, T.; Sun, Z.; Ozair, M.Z.; Tchieu, J.; Zimmer, B.; Fattahi, F.; et al. Combined small-molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nat. Biotechnol. 2017, 35, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Teng, J.; Takei, Y.; Oguchi, K.; Hirokawa, N. MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J. Cell Biol. 2002, 158, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.-Y.; Hu, Y.; Chen, C.; Yuan, F.; Xu, M.; Li, Q.; Fang, K.-H.; Chen, Y.; Liu, Y. Enhanced derivation of human pluripotent stem cell-derived cortical glutamatergic neurons by a small molecule. Sci. Rep. 2017, 7, 3282. [Google Scholar] [CrossRef]
- Du, Z.-W.; Chen, H.; Liu, H.; Lu, J.; Qian, K.; Huang, C.-L.; Zhong, X.; Fan, F.; Zhang, S.-C. Generation and Expansion of highly-pure Motor Neuron Progenitors from Human Pluripotent Stem Cells. Nat. Commun. 2015, 6, 6626. [Google Scholar] [CrossRef]
- Eiges, R.; Urbach, A.; Malcov, M.; Frumkin, T.; Schwartz, T.; Amit, A.; Yaron, Y.; Eden, A.; Yanuka, O.; Benvenisty, N.; et al. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 2007, 1, 568–577. [Google Scholar] [CrossRef]
- Biacsi, R.; Kumari, D.; Usdin, K. SIRT1 Inhibition Alleviates Gene Silencing in Fragile X Mental Retardation Syndrome. PLoS Genet. 2008, 4, e1000017. [Google Scholar] [CrossRef]
- Dolskiy, A.A.; Pustylnyak, V.O.; Yarushkin, A.A.; Lemskaya, N.A.; Yudkin, D.V. Inhibitors of Histone Deacetylases Are Weak Activators of the FMR1 Gene in Fragile X Syndrome Cell Lines. BioMed Res. Int. 2017, 2017, 3582601. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992.e6. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, J.G.; Henikoff, S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 2018, 13, 1006–1019. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Mancano, G.; Lanni, S.; Palumbo, F.; Goracci, M.; Ferrè, F.; Helmer-Citterich, M.; Neri, G. Genome-wide methylation analysis demonstrates that 5-aza-2-deoxycytidine treatment does not cause random DNA demethylation in fragile X syndrome cells. Epigenetics Chromatin 2016, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Jüttermann, R.; Li, E.; Jaenisch, R. Toxicity of 5-aza-2’-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc. Natl. Acad. Sci. USA 1994, 91, 11797–11801. [Google Scholar] [CrossRef]
- Jin, P.; Duan, R.; Qurashi, A.; Qin, Y.; Tian, D.; Rosser, T.C.; Liu, H.; Feng, Y.; Warren, S.T. Pur α Binds to rCGG Repeats and Modulates Repeat-Mediated Neurodegeneration in a Drosophila Model of Fragile X Tremor/Ataxia Syndrome. Neuron 2007, 55, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Freyermuth, F.; Tabet, R.; Tran, T.; He, F.; Ruffenach, F.; Alunni, V.; Moine, H.; Thibault, C.; Page, A.; et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013, 3, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Sofola, O.A.; Jin, P.; Qin, Y.; Duan, R.; Liu, H.; de Haro, M.; Nelson, D.L.; Botas, J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 2007, 55, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Vershkov, D.; Fainstein, N.; Suissa, S.; Golan-Lev, T.; Ben-Hur, T.; Benvenisty, N. FMR1 Reactivating Treatments in Fragile X iPSC-Derived Neural Progenitors In Vitro and In Vivo. Cell Rep. 2019, 26, 2531–2539.e4. [Google Scholar] [CrossRef]
- Graef, J.D.; Wu, H.; Ng, C.; Sun, C.; Villegas, V.; Qadir, D.; Jesseman, K.; Warren, S.T.; Jaenisch, R.; Cacace, A.; et al. Partial FMRP expression is sufficient to normalize neuronal hyperactivity in Fragile X neurons. Eur. J. Neurosci. 2020, 51, 2143–2157. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nur, O.; Caspi, I.; Benvenisty, N. Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell Biol. 2012, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Gazy, I.; Usdin, K. Pharmacological Reactivation of the Silenced FMR1 Gene as a Targeted Therapeutic Approach for Fragile X Syndrome. Brain Sci. 2019, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Izant, J.G.; McIntosh, J.R. Microtubule-associated proteins: A monoclonal antibody to MAP2 binds to differentiated neurons. Proc. Natl. Acad. Sci. USA 1980, 77, 4741–4745. [Google Scholar] [CrossRef]
- Crandall, J.E.; Jacobson, M.; Kosik, K.S. Ontogenesis of microtubule-associated protein 2 (MAP2) in embryonic mouse cortex. Dev. Brain Res. 1986, 28, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Johnson GV, W.; Jope, R.S. The role of microtubule-associated protein 2 (MAP-2) in neuronal growth, plasticity, and degeneration. J. Neurosci. Res. 1992, 33, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.R.; Pluciennik, A. DNA Mismatch Repair and its Role in Huntington’s Disease. J. Huntingt. Dis. 2021, 10, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Corless, S.; Gilbert, N. Effects of DNA supercoiling on chromatin architecture. Biophys. Rev. 2016, 8, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Nobile, V.; Pucci, C.; Chiurazzi, P. Mechanisms of the FMR1 Repeat Instability: How Does the CGG Sequence Expand? Int. J. Mol. Sci. 2022, 23, 5425. [Google Scholar] [CrossRef] [PubMed]
- Czuppa, M.; Dhingra, A.; Zhou, Q.; Schludi, C.; König, L.; Scharf, E.; Farny, D.; Dalmia, A.; Täger, J.; Castillo-Lizardo, M.; et al. Drug screen in iPSC-Neurons identifies nucleoside analogs as inhibitors of (G4C2)n expression in C9orf72 ALS/FTD. Cell Rep. 2022, 39, 110913. [Google Scholar] [CrossRef]
- Gu, X.; Tohme, R.; Tomlinson, B.; Sakre, N.; Hasipek, M.; Durkin, L.; Schuerger, C.; Grabowski, D.; Zidan, A.M.; Radivoyevitch, T.; et al. Decitabine- and 5-azacytidine resistance emerges from adaptive responses of the pyrimidine metabolism network. Leukemia 2021, 35, 1023–1036. [Google Scholar] [CrossRef]
- Almqvist, H.; Axelsson, H.; Jafari, R.; Dan, C.; Mateus, A.; Haraldsson, M.; Larsson, A.; Molina, D.M.; Artursson, P.; Lundbäck, T.; et al. CETSA screening identifies known and novel thymidylate synthase inhibitors and slow intracellular activation of 5-fluorouracil. Nat. Commun. 2016, 7, 11040. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kam, C.W.; Dumelie, J.G.; Ciceri, G.; Yang, W.-Y.; Disney, M.D.; Studer, L.; Jaffrey, S.R. Sustained Epigenetic Reactivation in Fragile X Neurons with an RNA-Binding Small Molecule. Genes 2025, 16, 278. https://doi.org/10.3390/genes16030278
Kam CW, Dumelie JG, Ciceri G, Yang W-Y, Disney MD, Studer L, Jaffrey SR. Sustained Epigenetic Reactivation in Fragile X Neurons with an RNA-Binding Small Molecule. Genes. 2025; 16(3):278. https://doi.org/10.3390/genes16030278
Chicago/Turabian StyleKam, Christina W., Jason G. Dumelie, Gabriele Ciceri, Wang-Yong Yang, Matthew D. Disney, Lorenz Studer, and Samie R. Jaffrey. 2025. "Sustained Epigenetic Reactivation in Fragile X Neurons with an RNA-Binding Small Molecule" Genes 16, no. 3: 278. https://doi.org/10.3390/genes16030278
APA StyleKam, C. W., Dumelie, J. G., Ciceri, G., Yang, W.-Y., Disney, M. D., Studer, L., & Jaffrey, S. R. (2025). Sustained Epigenetic Reactivation in Fragile X Neurons with an RNA-Binding Small Molecule. Genes, 16(3), 278. https://doi.org/10.3390/genes16030278