
Evaluation of Pan-Cancer Immune Heterogeneity Based on DNA Methylation


Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Cell Type-Specific DNA Methylation Genes Selection
2.3. Pan-Cancer Tissue Deconvolution
2.4. Clustering Based on Immune Infiltration
2.5. Analysis Based on Phenotypic Data
2.6. Analysis Based on Transcriptome
3. Results
3.1. Overview of the Study
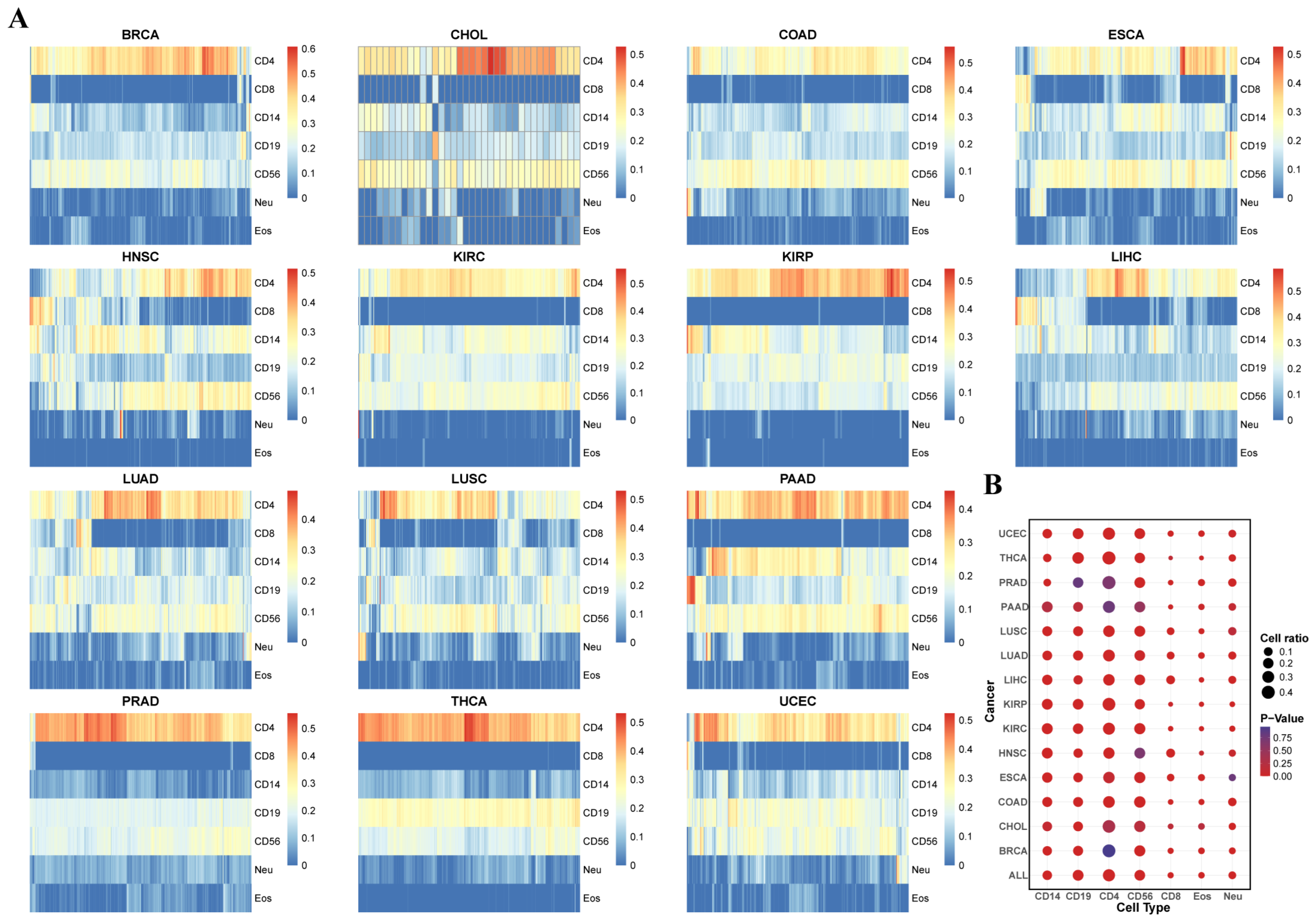
3.2. Evaluation of Tumor Microenvironment Immune Infiltration Based on Immune Cell Type-Specific Genes
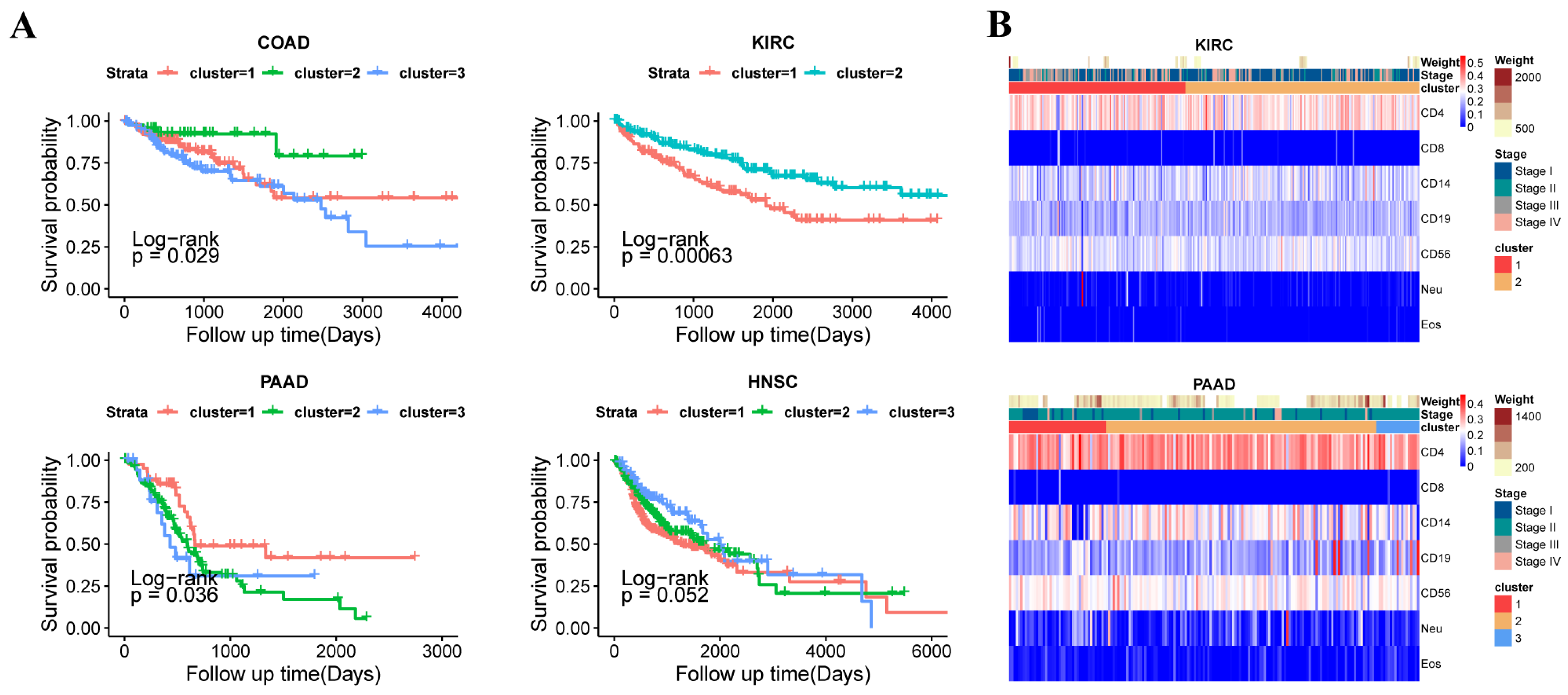
3.3. The Subtypes with Immune Infiltration Characteristics in 14 Tumors
3.4. The Different Subtypes Showed the Differential Phenotypes
3.5. The Analysis of Transcriptome Between Subtypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lei, X.; Lei, Y.; Li, J.-K.; Du, W.-X.; Li, R.-G.; Yang, J.; Li, J.; Li, F.; Tan, H.-B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Ge, R.; Wang, Z.; Cheng, L. Tumor microenvironment heterogeneity: An important mediator of prostate cancer progression and therapeutic resistance. NPJ Precis. Oncol. 2022, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, S.; Kim, J.; Kim, Y.; Yoon, H.M.; Rayhan, B.R.; Jeong, J.; Bothwell, A.L.M.; Shin, J.H. Trogocytosis-mediated immune evasion in the tumor microenvironment. Exp. Mol. Med. 2025, 57, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Shi, Y.; Hao, D.; Qian, H.; Tao, Z. Natural killer cell-based cancer immunotherapy: From basics to clinical trials. Exp. Hematol. Oncol. 2024, 13, 101. [Google Scholar] [CrossRef]
- Luyang, H.; Zeng, F.; Lei, Y.; He, Q.; Zhou, Y.; Xu, J. Bidirectional role of neutrophils in tumor development. Mol. Cancer 2025, 24, 1–16. [Google Scholar] [CrossRef]
- Elguindy, M.M.; Young, J.S.; Ho, W.S.; Lu, R.O. Co-evolution of glioma and immune microenvironment. J. Immunother. Cancer 2024, 12, e009175. [Google Scholar] [CrossRef] [PubMed]
- Ucche, S.; Hayakawa, Y. Immunological Aspects of Cancer Cell Metabolism. Int. J. Mol. Sci. 2024, 25, 5288. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.A.; Neganova, M.E.; Aleksandrova, Y.; Burcher, J.T.; Chugunova, E.; Fan, R.; Tse, E.; Sethi, G.; Bishayee, A.; Liu, J. Signaling controversy and future therapeutic perspectives of targeting sphingolipid network in cancer immune editing and resistance to tumor necrosis factor-α immunotherapy. Cell Commun. Signal. 2024, 22, 251. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Liu, Z.; Tan, X.; Wang, G.; Xu, Y.; Xie, L.; Lin, Y. Application of immune cell infiltration in the diagnosis and prognosis of non-small cell lung cancer. Chin. J. Biotechnol. 2020, 36, 740–749. [Google Scholar]
- Pikul, J.; Machnicki, M.M.; Rzepakowska, A.; Winiarska, N.; Chudy, A.; Moskowicz, A.; Król, K.; Fus, Ł.; Kostrzewa, G.; Stokłosa, T. Potentially actionable molecular alterations related to poor oncologic outcomes in salivary gland carcinomas. BMC Cancer 2025, 25, 42. [Google Scholar] [CrossRef]
- Kim, J.H.; Hong, J.; Lee, J.A.; Jung, M.; Choi, E.; Cho, N.-Y.; Kang, G.H.; Kim, S. Immune microenvironmental heterogeneity according to tumor DNA methylation phenotypes in microsatellite instability-high colorectal cancers. Cancer Immunol. Immunother. 2024, 73, 215. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Loyfer, N.; Magenheim, J.; Peretz, A.; Cann, G.; Bredno, J.; Klochendler, A.; Fox-Fisher, I.; Shabi-Porat, S.; Hecht, M.; Pelet, T.; et al. A DNA methylation atlas of normal human cell types. Nature 2023, 613, 355–364. [Google Scholar] [CrossRef]
- Ting, A.H.; Reinius, L.E.; Acevedo, N.; Joerink, M.; Pershagen, G.; Dahlén, S.-E.; Greco, D.; Söderhäll, C.; Scheynius, A.; Kere, J. Differential DNA methylation in purified human blood cells: Implications for cell lineage and studies on disease susceptibility. PLoS ONE 2012, 7, e41361. [Google Scholar]
- Zhang, Y.; Liu, H.; Lv, J.; Xiao, X.; Zhu, J.; Liu, X.; Su, J.; Li, X.; Wu, Q.; Wang, F.; et al. QDMR: A quantitative method for identification of differentially methylated regions by entropy. Nucleic Acids Res. 2011, 39, e58. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C. The mathematical theory of communication. MD Comput. 1997, 14, 306–317. [Google Scholar] [PubMed]
- Chakravarthy, A.; Furness, A.; Joshi, K.; Ghorani, E.; Ford, K.; Ward, M.J.; King, E.V.; Lechner, M.; Marafioti, T.; Quezada, S.A.; et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat. Commun. 2018, 9, 3220. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, D.; Idnani, A. Dual and primal-dual methods for solving strictly convex quadratic programs. Numer. Anal. 1982, 909, 226–239. [Google Scholar]
- Goldfarb, D.; Idnani, A. A numerically stable dual method for solving strictly convex quadratic programs. Math. Program. 1983, 27, 1–33. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Dieci, M.V.; Miglietta, F.; Guarneri, V. Immune infiltrates in breast cancer: Recent updates and clinical implications. Cells 2021, 10, 226. [Google Scholar] [CrossRef]
- Huang, C.; Luo, M.-Y.; Wen, N.-Q.; Chen, Y.-M.; Zhang, L.-Z.; Cao, Y. The prognostic implications and oncogenic role of NSUN5 in clear cell renal cell carcinoma. Clin. Exp. Med. 2024, 25, 8. [Google Scholar] [CrossRef]
- Xiang, H.; Kasajima, R.; Azuma, K.; Tagami, T.; Hagiwara, A.; Nakahara, Y.; Saito, H.; Igarashi, Y.; Wei, F.; Ban, T.; et al. Multi-omics analysis-based clinical and functional significance of a novel prognostic and immunotherapeutic gene signature derived from amino acid metabolism pathways in lung adenocarcinoma. Front. Immunol. 2024, 15, 1361992. [Google Scholar] [CrossRef]
- Li, D.; Cao, D.; Zhang, Y.; Yu, X.; Wu, Y.; Jia, Z.; Jiang, J.; Cao, X. Integrative pan-cancer analysis and experiment validation identified GLS as a biomarker in tumor progression, prognosis, immune microenvironment, and immunotherapy. Sci. Rep. 2025, 15, 525. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.-F.; Liu, C.-J.; Liu, L.-L.; Zhang, Q.; Guo, A.-Y. Expression profile of immune checkpoint genes and their roles in predicting immunotherapy response. Brief. Bioinform. 2021, 22, bbaa176. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Wu, L.; Jian, H.; Zhang, C.; Lou, Y.; Kang, Y.; Hou, M.; Li, Z.; Li, X.; Sun, B.; et al. Identification and characterization of aging/senescence-induced genes in osteosarcoma and predicting clinical prognosis. Front. Immunol. 2022, 13, 997765. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Breeze, C.E.; Zheng, S.C.; Beck, S. A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinform. 2017, 18, 1–14. [Google Scholar] [CrossRef]
- Wen, Y.; Wei, Y.; Zhang, S.; Li, S.; Liu, H.; Wang, F.; Zhao, Y.; Zhang, D.; Zhang, Y. Cell subpopulation deconvolution reveals breast cancer heterogeneity based on DNA methylation signature. Brief. Bioinform. 2016, 18, 426–440. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Tumor/Cell-Types | Number of Samples |
|---|---|---|
| TCGA | BRCA | 794 |
| CHOL | 45 | |
| COAD | 309 | |
| ESCA | 186 | |
| HNSC | 530 | |
| KIRC | 323 | |
| KIRP | 276 | |
| LIHC | 380 | |
| LUAD | 471 | |
| LUSC | 370 | |
| PAAD | 185 | |
| PRAD | 503 | |
| THCA | 515 | |
| UCEC | 436 | |
| GEO | CD4 | 6 |
| CD8 | 6 | |
| CD14 | 6 | |
| CD19 | 6 | |
| CD56 | 6 | |
| Neu | 6 | |
| Eos | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Liu, J.; Shi, B.; Ma, T.; Yu, P.; Li, J.; Gu, Y.; Zhang, Y. Evaluation of Pan-Cancer Immune Heterogeneity Based on DNA Methylation. Genes 2025, 16, 160. https://doi.org/10.3390/genes16020160
Zhou Y, Liu J, Shi B, Ma T, Yu P, Li J, Gu Y, Zhang Y. Evaluation of Pan-Cancer Immune Heterogeneity Based on DNA Methylation. Genes. 2025; 16(2):160. https://doi.org/10.3390/genes16020160
Chicago/Turabian StyleZhou, Yang, Jiebiao Liu, Bowen Shi, Te Ma, Peishen Yu, Ji Li, Yue Gu, and Yan Zhang. 2025. "Evaluation of Pan-Cancer Immune Heterogeneity Based on DNA Methylation" Genes 16, no. 2: 160. https://doi.org/10.3390/genes16020160
APA StyleZhou, Y., Liu, J., Shi, B., Ma, T., Yu, P., Li, J., Gu, Y., & Zhang, Y. (2025). Evaluation of Pan-Cancer Immune Heterogeneity Based on DNA Methylation. Genes, 16(2), 160. https://doi.org/10.3390/genes16020160