
APCAD Part 2: A Novel Method for Detection of Meiotic Aneuploidy in Preimplantation Embryos


Abstract
1. Introduction
2. Materials and Methods
2.1. IVF Treatment and Embryo Manipulations
2.2. Genomic DNA and Embryos
2.3. WGA and SNP Array
3. Results
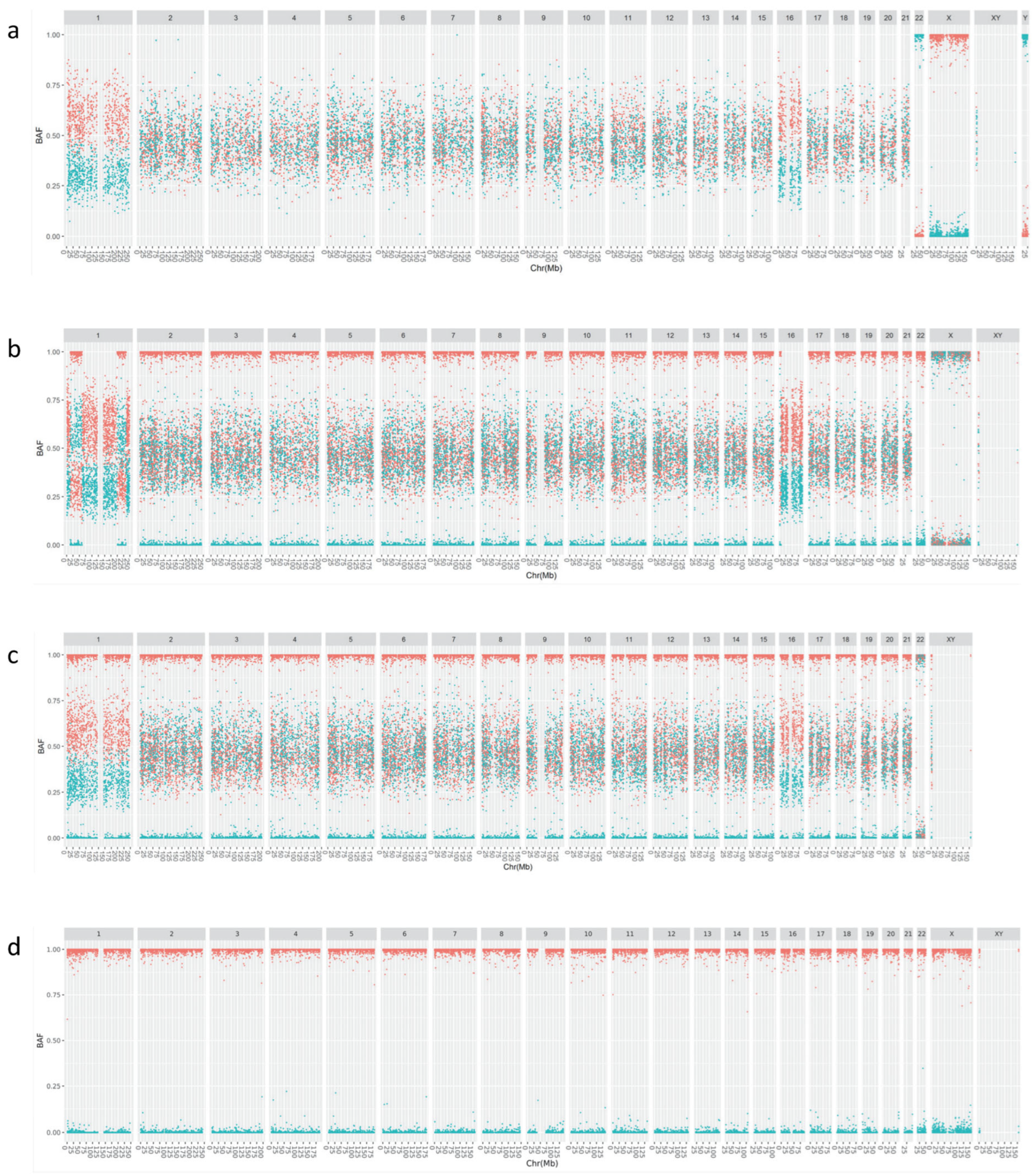
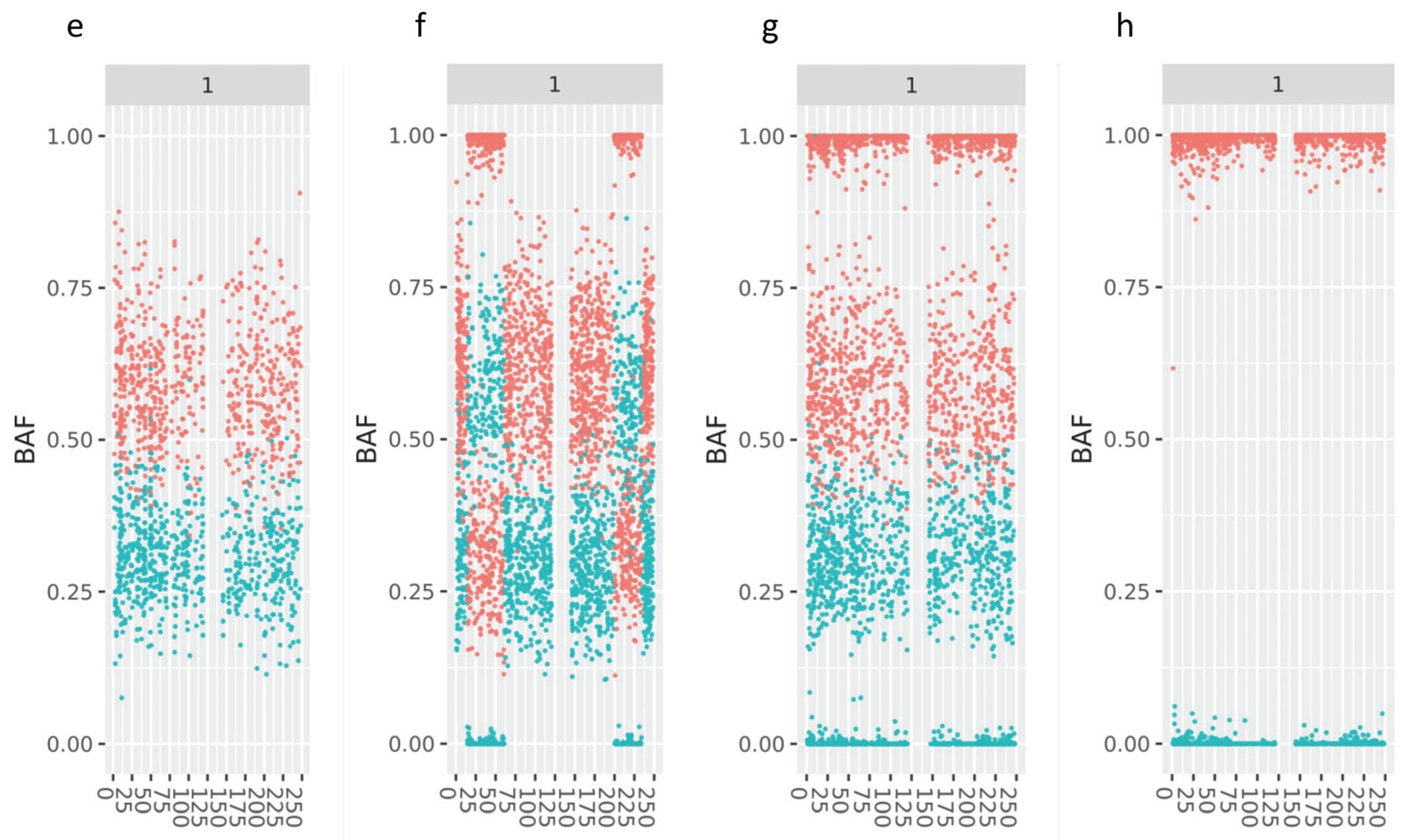
3.1. Improved Visualization Using cBAF Profiles
3.2. Retrospective Analysis Using APCAD Profiles
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Natesan, S.A.; Handyside, A.H.; Thornhill, A.R.; Ottolini, C.S.; Sage, K.; Summers, M.C.; Konstantinidis, M.; Wells, D.; Griffin, D.K. Live Birth after PGD with Confirmation by a Comprehensive Approach (Karyomapping) for Simultaneous Detection of Monogenic and Chromosomal Disorders. Reprod. Biomed. Online 2014, 29, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Natesan, S.A.; Bladon, A.J.; Coskun, S.; Qubbaj, W.; Prates, R.; Munne, S.; Coonen, E.; Dreesen, J.C.F.M.; Stevens, S.J.C.; Paulussen, A.D.C.; et al. Genome-Wide Karyomapping Accurately Identifies the Inheritance of Single-Gene Defects in Human Preimplantation Embryos in Vitro. Genet. Med. Off. J. Am. Coll. Med. Genet. 2014, 16, 838–845. [Google Scholar] [CrossRef]
- Beyer, C.E.; Lewis, A.; Willats, E.; Mullen, J. Preimplantation Genetic Testing Using Karyomapping for a Paternally Inherited Reciprocal Translocation: A Case Study. J. Assist. Reprod. Genet. 2019, 36, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nagi, J.; Jones, B.; Naja, R.; Amer, A.; Sunkara, S.; SenGupta, S.; Serhal, P. Live Birth Rate Is Associated with Oocyte Yield and Number of Biopsied and Suitable Blastocysts to Transfer in Preimplantation Genetic Testing (PGT) Cycles for Monogenic Disorders and Chromosomal Structural Rearrangements. Eur. J. Obstet. Gynecol. Reprod. Biol. X 2019, 4, 100055. [Google Scholar] [CrossRef] [PubMed]
- Samarasekera, T.; Willats, E.; Green, M.P.; Hardy, T.; Rombauts, L.; Zander-Fox, D. Impact of Male Age on Paternal Aneuploidy: Single-Nucleotide Polymorphism Microarray Outcomes Following Blastocyst Biopsy. Reprod. Biomed. Online 2023, 47, 103245. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, D.; Hornak, M.; Horak, J.; Navratil, R.; Tauwinklova, G.; Rubes, J.; Vesela, K. Incidence and Origin of Meiotic Whole and Segmental Chromosomal Aneuploidies Detected by Karyomapping. Reprod. Biomed. Online 2019, 38, 330–339. [Google Scholar] [CrossRef]
- Rabinowitz, M.; Ryan, A.; Gemelos, G.; Hill, M.; Baner, J.; Cinnioglu, C.; Banjevic, M.; Potter, D.; Petrov, D.A.; Demko, Z. Origins and Rates of Aneuploidy in Human Blastomeres. Fertil. Steril. 2012, 97, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Verdyck, P.; Berckmoes, V.; Van Laere, S.; Keymolen, K.; Olsen, C.; De Rycke, M. Analysis of Parental Contribution for Aneuploidy Detection (APCAD): A Novel Method to Detect Aneuploidy and Mosaicism in Preimplantation Embryos. Reprod. Biomed. Online 2022, 44, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Viotti, M.; Victor, A.R.; Barnes, F.L.; Zouves, C.G.; Besser, A.G.; Grifo, J.A.; Cheng, E.-H.; Lee, M.-S.; Horcajadas, J.A.; Corti, L.; et al. Using Outcome Data from One Thousand Mosaic Embryo Transfers to Formulate an Embryo Ranking System for Clinical Use. Fertil. Steril. 2021, 115, 1212–1224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Chen, J.J.; Nabu, S.; Yeung, Q.S.Y.; Li, Y.; Tan, J.H.; Suksalak, W.; Chanchamroen, S.; Quangkananurug, W.; Wong, P.S.; et al. The Pregnancy Outcome of Mosaic Embryo Transfer: A Prospective Multicenter Study and Meta-Analysis. Genes 2020, 11, 973. [Google Scholar] [CrossRef]
- Capalbo, A.; Poli, M.; Rienzi, L.; Girardi, L.; Patassini, C.; Fabiani, M.; Cimadomo, D.; Benini, F.; Farcomeni, A.; Cuzzi, J.; et al. Mosaic Human Preimplantation Embryos and Their Developmental Potential in a Prospective, Non-Selection Clinical Trial. Am. J. Hum. Genet. 2021, 108, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- De Witte, L.; Baetens, M.; Tilleman, K.; Vanden Meerschaut, F.; Janssens, S.; Van Tongerloo, A.; Szymczak, V.; Stoop, D.; Dheedene, A.; Symoens, S.; et al. Aligning Genotyping and Copy Number Data in Single Trophectoderm Biopsies for Aneuploidy Prediction: Uncovering Incomplete Concordance. Hum. Reprod. Open 2024, 2024, hoae056. [Google Scholar] [CrossRef]
- Janssen, A.E.J.; Koeck, R.M.; Essers, R.; Cao, P.; van Dijk, W.; Drüsedau, M.; Meekels, J.; Yaldiz, B.; van de Vorst, M.; de Koning, B.; et al. Clinical-Grade Whole Genome Sequencing-Based Haplarithmisis Enables All Forms of Preimplantation Genetic Testing. Nat. Commun. 2024, 15, 7164. [Google Scholar] [CrossRef] [PubMed]
- De Rycke, M.; De Vos, A.; Belva, F.; Berckmoes, V.; Bonduelle, M.; Buysse, A.; Keymolen, K.; Liebaers, I.; Nekkebroeck, J.; Verdyck, P.; et al. Preimplantation Genetic Testing with HLA Matching: From Counseling to Birth and Beyond. J. Hum. Genet. 2020, 65, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, C.S.; Rogers, S.; Sage, K.; Summers, M.C.; Capalbo, A.; Griffin, D.K.; Sarasa, J.; Wells, D.; Handyside, A.H. Karyomapping Identifies Second Polar Body DNA Persisting to the Blastocyst Stage: Implications for Embryo Biopsy. Reprod. Biomed. Online 2015, 31, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Zamani Esteki, M.; Dimitriadou, E.; Mateiu, L.; Melotte, C.; Van der Aa, N.; Kumar, P.; Das, R.; Theunis, K.; Cheng, J.; Legius, E.; et al. Concurrent Whole-Genome Haplotyping and Copy-Number Profiling of Single Cells. Am. J. Hum. Genet. 2015, 96, 894–912. [Google Scholar] [CrossRef] [PubMed]
- Barad, D.H.; Albertini, D.F.; Molinari, E.; Gleicher, N. IVF Outcomes of Embryos with Abnormal PGT-A Biopsy Previously Refused Transfer: A Prospective Cohort Study. Hum. Reprod. Oxf. Engl. 2022, 37, 1194–1206. [Google Scholar] [CrossRef]
- Tiegs, A.W.; Tao, X.; Zhan, Y.; Whitehead, C.; Kim, J.; Hanson, B.; Osman, E.; Kim, T.J.; Patounakis, G.; Gutmann, J.; et al. A Multicenter, Prospective, Blinded, Nonselection Study Evaluating the Predictive Value of an Aneuploid Diagnosis Using a Targeted next-Generation Sequencing-Based Preimplantation Genetic Testing for Aneuploidy Assay and Impact of Biopsy. Fertil. Steril. 2021, 115, 627–637. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Categories | Paternal Genotype Call | Maternal Genotype Call | Expected Genotype Sample * | Remark |
|---|---|---|---|---|
| Category 1 | ||||
| Subcategory 1A | BB | AA | AB | |
| BB | NC | NC or BB | Only for SNPs on Y chr. | |
| Subcategory 1B | AA | BB | AB | |
| AA | NC | NC or AA | Only for SNPs on Y chr. | |
| Category 2 | ||||
| Subcategory 2A | BB | AB | AB or BB | Not phased |
| Subcategory 2B | AA | AB | AA or AB | Not phased |
| Category 3 | ||||
| Subcategory 3A | AB | BB | AB or BB | Not phased |
| Subcategory 3B | AB | AA | AA or AB | Not phased |
| Category 4 | ||||
| Subcategory 4A | BB | BB | BB | |
| Subcategory 4B | AA | AA | AA |
| Type of Anomaly | N of Mat. Origin | N of Pat Origin | Remark |
|---|---|---|---|
| De novo abnormalities (PGT-A) | |||
| BPH trisomy autosome | 56 | 1 | |
| BPH trisomy XXY | 3 | 2 | |
| BPH segmental CN gain autosome | 1 | 0 | Sized 84 Mb |
| BPH segmental CN gain and deletion on autosome | 1 | 0 | Isochromosome 9q (deletion of 9p and BPH copy number gain of 9q) |
| Monosomy autosome | 53 | 3 | |
| Monosomy X | 0 | 6 | |
| Deletion on autosome | 2 | 23 | Maternal: sized 13 and 84 Mb. Paternal: sized 10, 14, 19, 25, 27, 30, 31, 32, 34, 40, 43, 47, 50, 51, 65, 65, 70, 78, 78, 81, 93, 103 and 123 Mb. |
| Deletion on X | 1 | 0 | Sized 58 Mb. |
| Inherited abnormalities (PGT-SR) | |||
| BPH tetrasomy autosome | 1 | 0 | |
| BPH trisomy autosome | 2 | 0 | |
| BPH segmental CN gain autosome | 2 | 0 | Sized 58 and 77 Mb |
| Monosomy autosome | 1 | 0 | |
| Deletion on autosome | 2 | 0 | Sized 11 and 29 Mb |
| Interpretation Karyomapping | Interpretation APCAD | |
|---|---|---|
| 1 | BPH CN gain 17q21.34 to qter (2× mat) | BPH CN gain 17q21.34 to qter (3× mat) (~38 Mb) |
| 2 | Normal disomy | BPH CN gain 1q23.3 to q24.2 (2× mat; ~6 Mb), estimated mosaic |
| 3 | Mosaic monosomy chr10 in the majority of the analyzed cells | Pat deletion of 10q23.1 to 10q24.31 (~20 Mb) and mosaic CN loss of remainder of chr10 in the majority of the analyzed cells * |
| 4 | Mosaic monosomy chr3 in the majority of the analyzed cells | Pat deletion of pter to 3p24.2 (~25 Mb) and mosaic CN loss of remainder of chr3 in the majority of the analyzed cells * |
| 5 | Monosomy chr17 pat | Pat deletion of 17q12 to qter (~49 Mb) and mosaic CN loss of the remainder of chr17 in the majority of the analyzed cells * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdyck, P.; Berckmoes, V.; Fernandez Gallardo, E.; Keymolen, K.; Olsen, C.; De Rycke, M. APCAD Part 2: A Novel Method for Detection of Meiotic Aneuploidy in Preimplantation Embryos. Genes 2025, 16, 115. https://doi.org/10.3390/genes16020115
Verdyck P, Berckmoes V, Fernandez Gallardo E, Keymolen K, Olsen C, De Rycke M. APCAD Part 2: A Novel Method for Detection of Meiotic Aneuploidy in Preimplantation Embryos. Genes. 2025; 16(2):115. https://doi.org/10.3390/genes16020115
Chicago/Turabian StyleVerdyck, Pieter, Veerle Berckmoes, Elia Fernandez Gallardo, Kathelijn Keymolen, Catharina Olsen, and Martine De Rycke. 2025. "APCAD Part 2: A Novel Method for Detection of Meiotic Aneuploidy in Preimplantation Embryos" Genes 16, no. 2: 115. https://doi.org/10.3390/genes16020115
APA StyleVerdyck, P., Berckmoes, V., Fernandez Gallardo, E., Keymolen, K., Olsen, C., & De Rycke, M. (2025). APCAD Part 2: A Novel Method for Detection of Meiotic Aneuploidy in Preimplantation Embryos. Genes, 16(2), 115. https://doi.org/10.3390/genes16020115