
Transcriptional Analysis of Tissues in Tartary Buckwheat Seedlings Under IAA Stimulation

Abstract
1. Introduction
2. Material and Methods
2.1. Plant Material and Cultivation Control
2.2. Auxin (IAA) Treatments and Material Preparation
2.3. High Throughput Transcriptome Sequencing
2.4. Sequencing Data Analysis and Gene Expression Level Identification
2.5. Comparison of Gene Expression Between qPCR and Transcriptome Analysis
2.6. Identification and Analysis of Differentially Expressed Genes (DEGs)
2.7. Identification and Expession Analysis of Auxin Signaling Pathway Genes
2.8. Data Availability Statement
3. Results
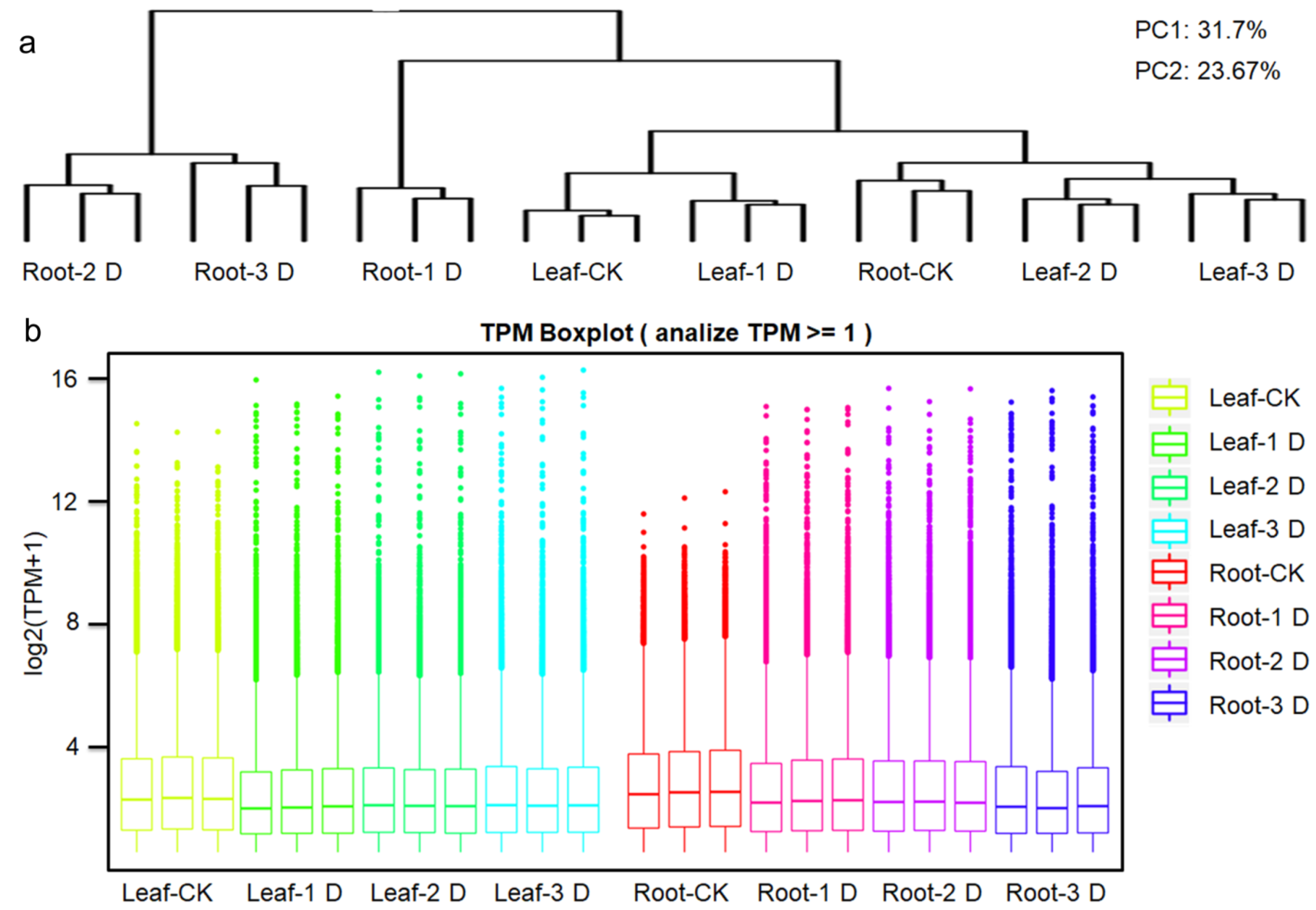
3.1. Statistics of Expressed Genes Through High-Throughput Transcriptome Analysis
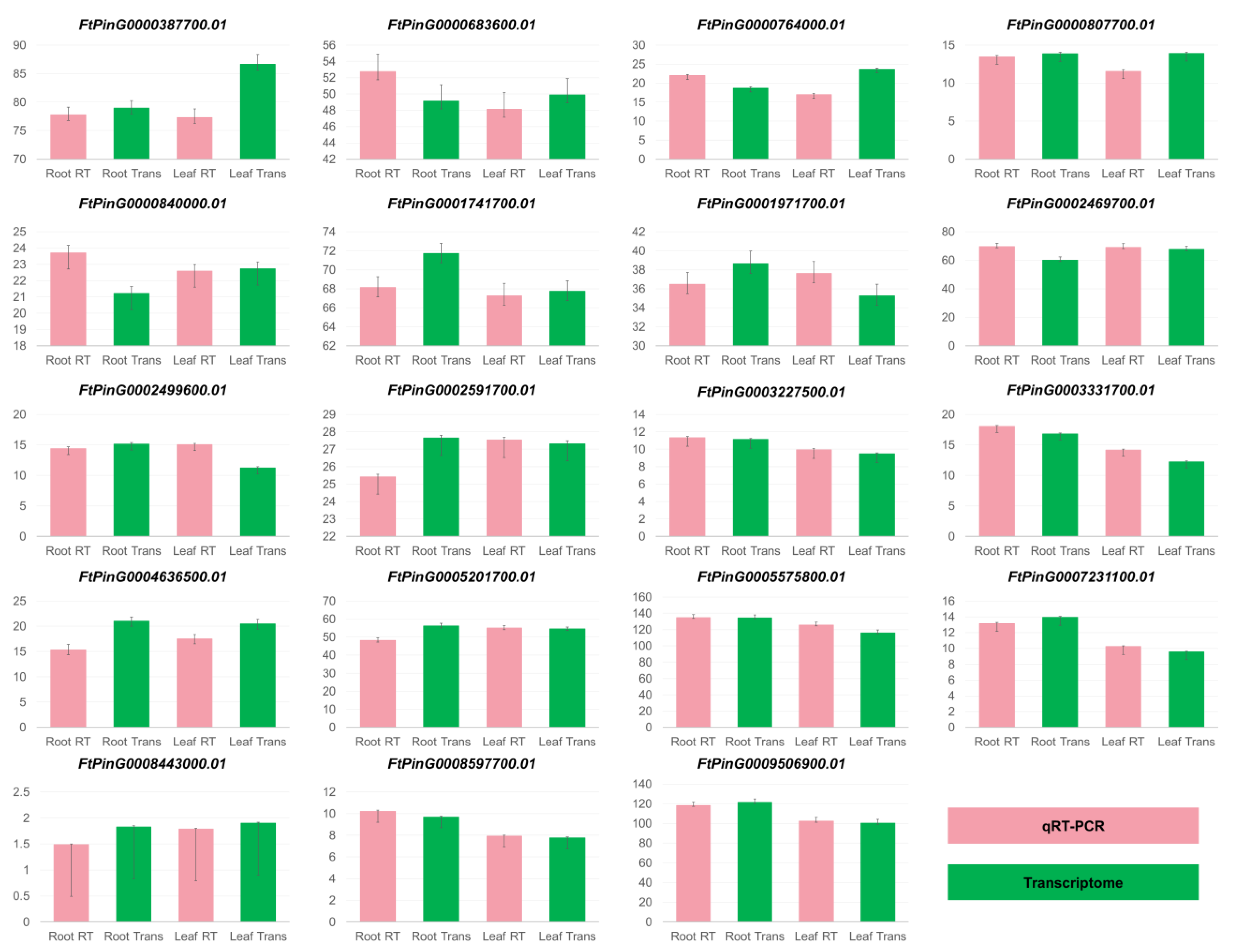
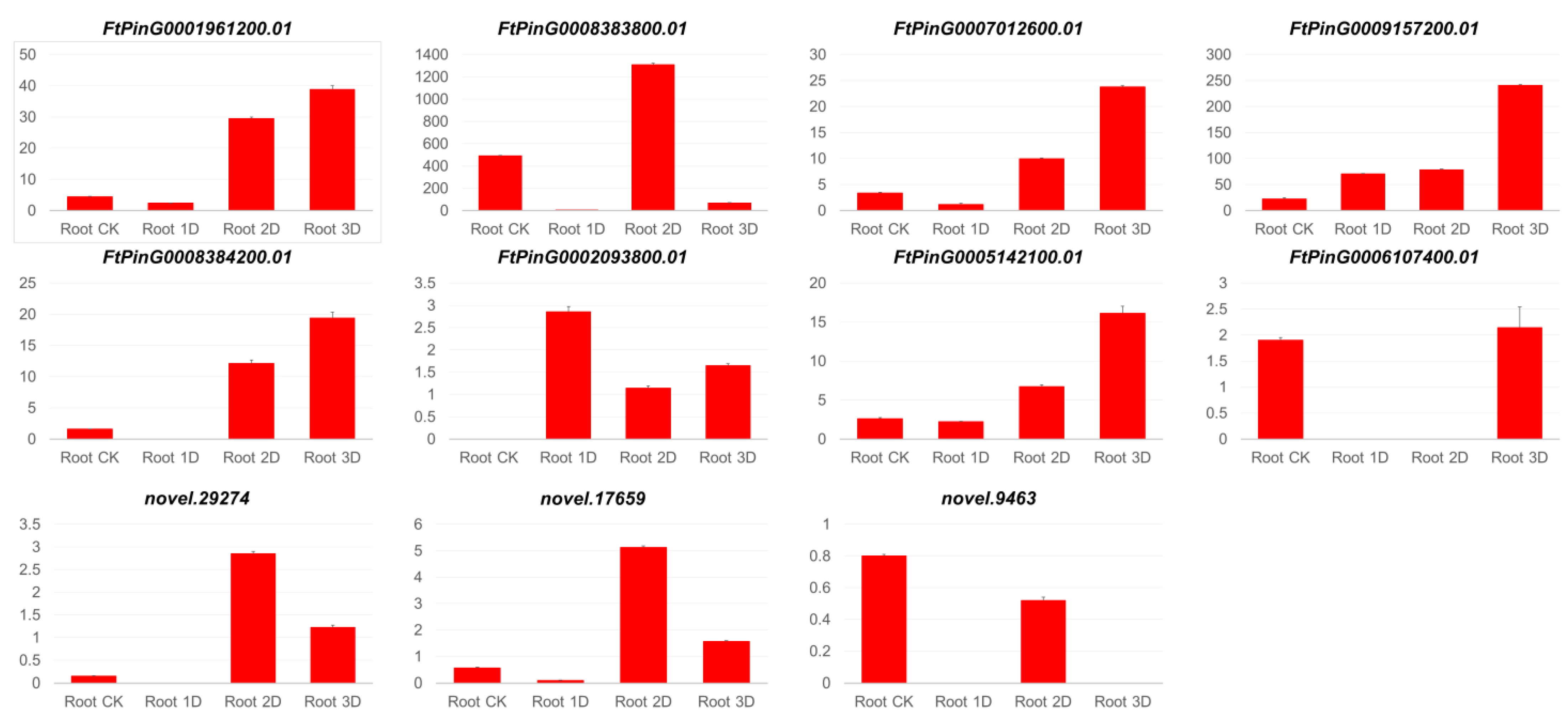
3.2. Comparison of Gene Expression Between qRT-PCR and Transcriptome Analysis
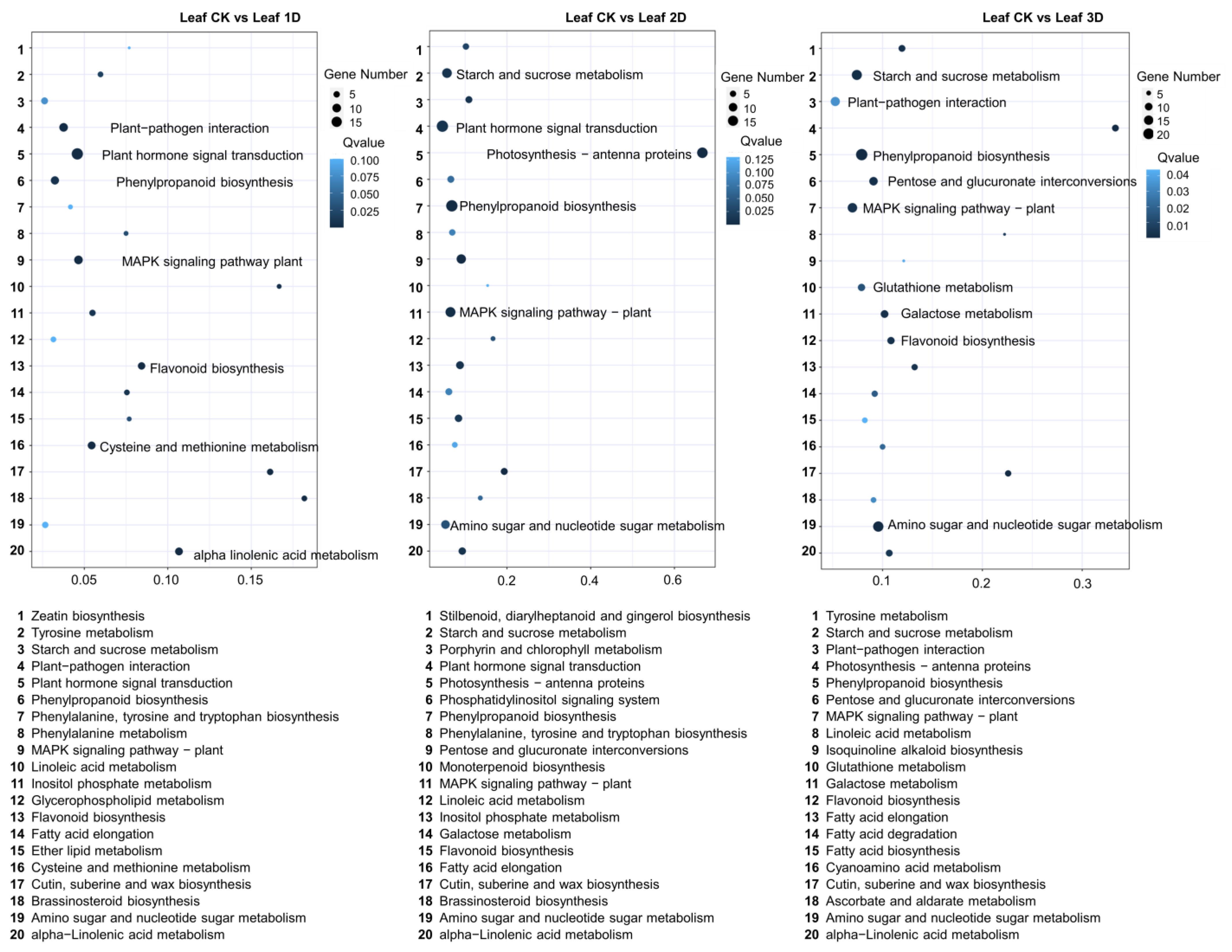
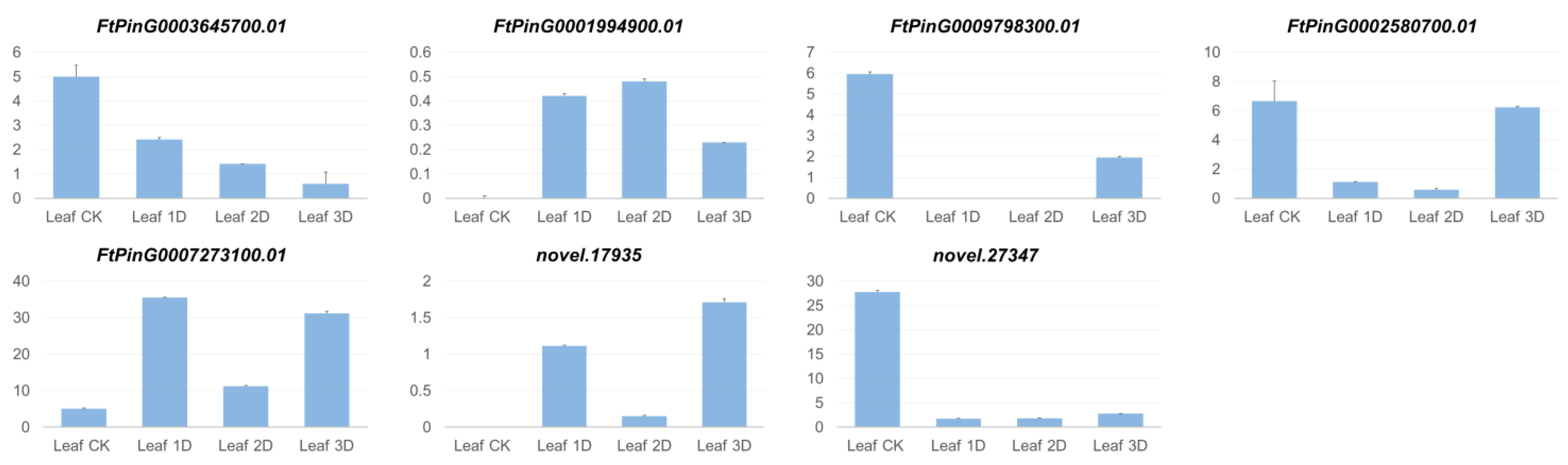
3.3. Identification of Differentially Expressed Genes (DEGs) in Leaf
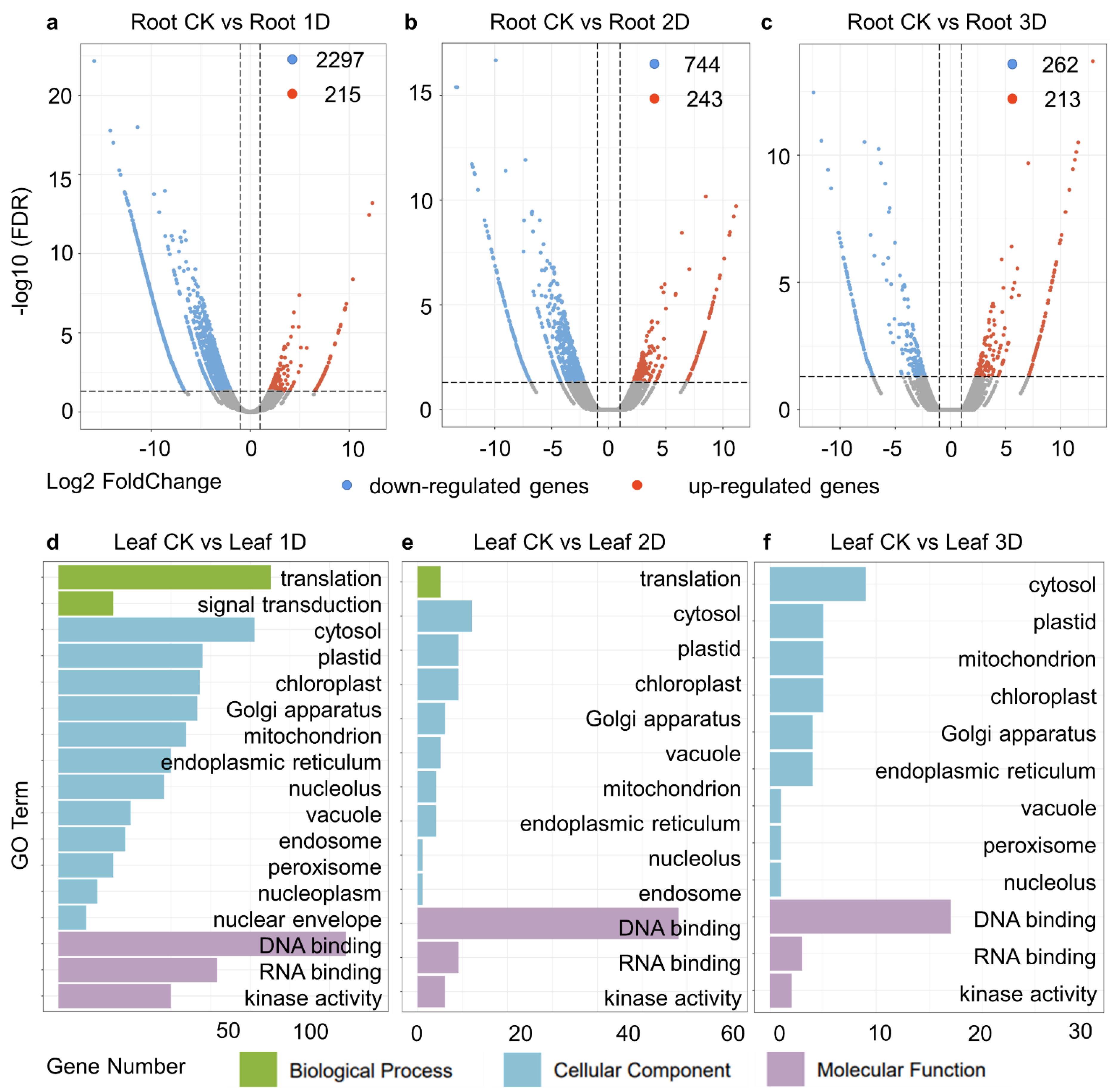
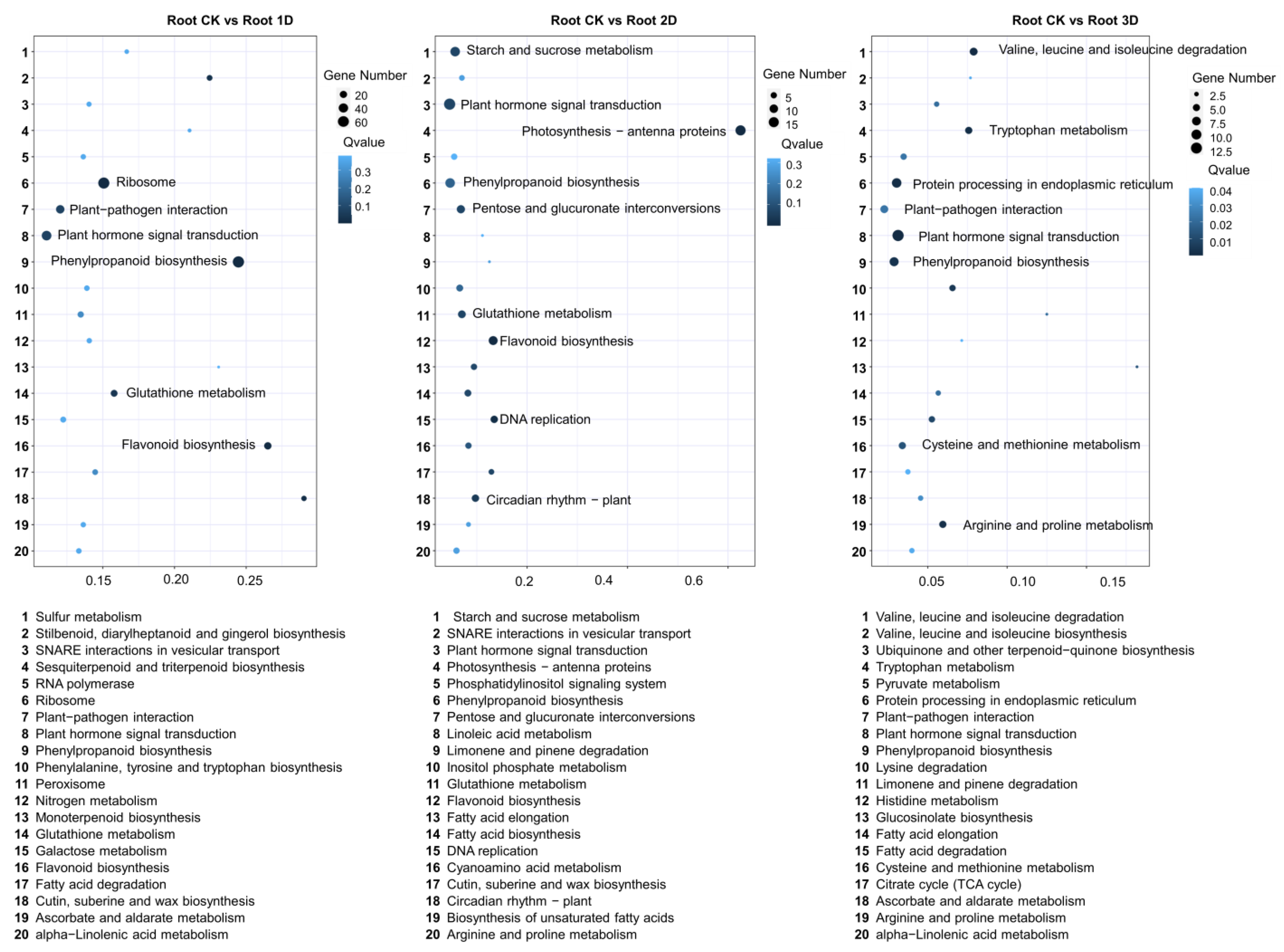
3.4. Identification of Differentially Expressed Genes (DEGs) in Root
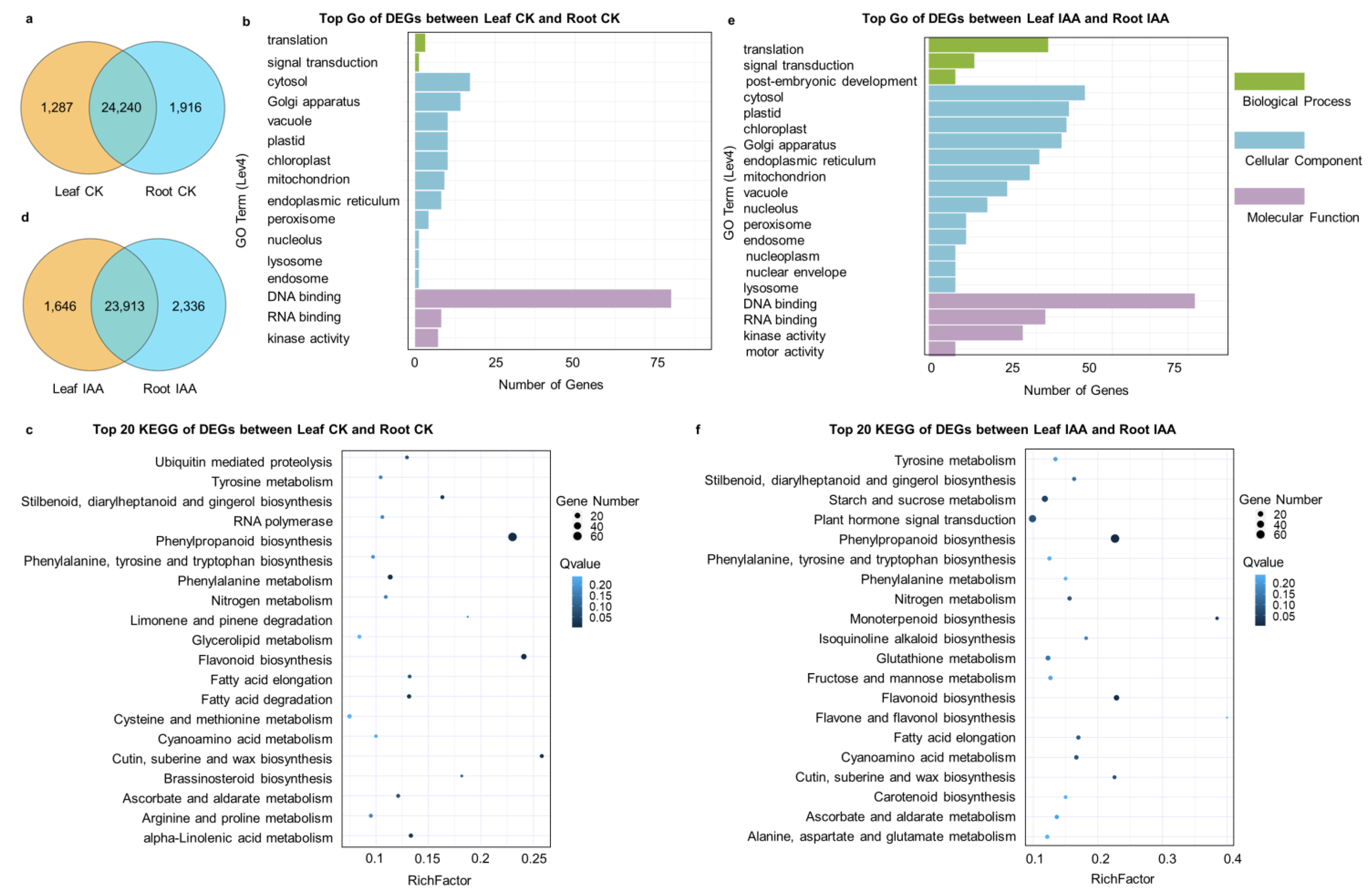
3.5. Specifically Expressed Genes in Leaves and Roots, as Well as DEGs in Various Tissues
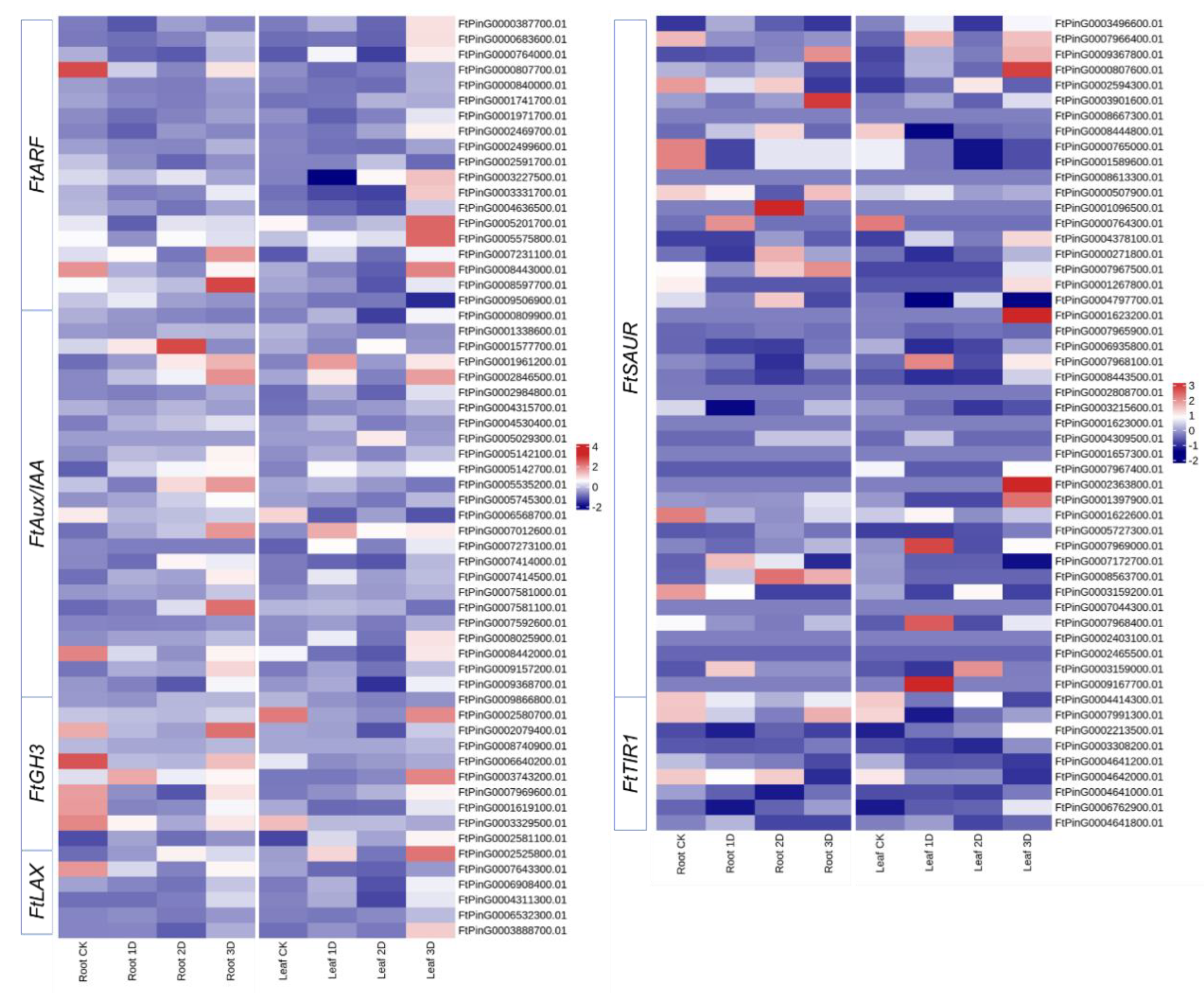
3.6. Identification and Expression Analysis of Auxin Signaling Pathway Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, Z.; Zhang, F.; Friml, J.; Ding, Z. Auxin signaling: Research advances over the past 30 years. J. Integr. Plant Biol. 2022, 64, 371–392. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.Y.; Dodd, I.C.; Chen, J.E.; Phang, S.M.; Chin, C.F.; Yow, Y.Y.; Ratnayeke, S. Regulation of algal and cyanobacterial auxin production, physiology, and application in agriculture: An overview. J. Appl. Phycol. 2021, 33, 2995–3023. [Google Scholar] [CrossRef]
- Du, M.; Spalding, E.P.; Gray, W.M. Rapid auxin-mediated cell expansion. Annu. Rev. Plant Biol. 2020, 71, 379–402. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Corrionero, A.; Sánchez-Vicente, I.; Arteaga, N.; Manrique-Gil, I.; Gómez-Jiménez, S.; Torres-Quezada, I.; Albertos, P.; Lorenzo, O. Fine-tuned nitric oxide and hormone interface in plant root development and regeneration. J. Exp. Bot. 2023, 74, 6104–6118. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, J.; Xu, X.; Wang, R.; Liu, Y.; Huang, S.; Wei, H.; Wei, Z. Molecular mechanisms of diverse auxin responses during plant growth and development. Int. J. Mol. Sci. 2022, 23, 12495. [Google Scholar] [CrossRef]
- Zhang, Y.; Berman, A.; Shani, E. Plant hormone transport and localization: Signaling molecules on the move. Annu. Rev. Plant Biol. 2023, 74, 453–479. [Google Scholar] [CrossRef]
- Kaniganti, S.; Bhattacharya, J.; Petla, B.P.; Reddy, P.S. Strigolactone, a neglected plant hormone, with a great potential for crop improvement: Crosstalk with other plant hormones. Environ. Exp. Bot. 2022, 204, 105072. [Google Scholar] [CrossRef]
- Gomes, G.L.B.; Scortecci, K.C. Auxin and its role in plant development: Structure, signalling, regulation and response mechanisms. Plant Biol. 2021, 23, 894–904. [Google Scholar] [CrossRef]
- Batista-Silva, W.; de Paiva Gonçalves, J.; Siqueira, J.A.; Martins, A.O.; Ribeiro, D.M.; Nunes-Nesi, A.; Zsögön, A.; Araújo, W.L. Auxin metabolism and the modulation of plant growth. Environ. Exp. Bot. 2024, 226, 105917. [Google Scholar] [CrossRef]
- Huang, X.; Lu, Z.; Zhai, L.; Li, N.; Yan, H. The small auxin-up RNA SAUR10 is involved in the promotion of seedling growth in rice. Plants 2023, 12, 3880. [Google Scholar] [CrossRef]
- Yuan, H.; Cheng, M.; Fan, F.; Zheng, X.; Wang, R.; Si, F.; Luo, X.; Li, N.; Li, S. OsGRF6-OsYUCCA1/OsWRKY82 Signaling Cascade Upgrade Grain Yield and Bacterial Blight Resistance in Rice. Adv. Sci. 2024, 11, 2407733. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kamiya, N.; Morinaka, Y.; Matsuoka, M.; Sazuka, T. Auxin biosynthesis by the YUCCA genes in rice. Plant Physiol. 2007, 143, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; You, J.; Xiong, L. Characterization of OsIAA1 gene, a member of rice Aux/IAA family involved in auxin and brassinosteroid hormone responses and plant morphogenesis. Plant Mol. Biol. 2009, 70, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Kawakatsu, T.; Taramino, G.; Itoh, J.I.; Allen, J.; Sato, Y.; Hong, S.K.; Yule, R.; Nagasawa, N.; Kojima, M.; Kusaba, M.; et al. PLASTOCHRON3/GOLIATH encodes a glutamate carboxypeptidase required for proper development in rice. Plant J. 2009, 58, 1028–1040. [Google Scholar] [CrossRef]
- Kumar, M.; Le, D.T.; Hwang, S.; Seo, P.J.; Kim, H.U. Role of the INDETERMINATE DOMAIN genes in plants. Int. J. Mol. Sci. 2019, 20, 2286. [Google Scholar] [CrossRef]
- Cassani, E.; Villa, D.; Durante, M.; Landoni, M.; Pilu, R. The brachytic 2 and 3 maize double mutant shows alterations in plant growth and embryo development. Plant Growth Regul. 2011, 64, 185–192. [Google Scholar] [CrossRef]
- Landoni, M.; Cassani, E.; Ghidoli, M.; Colombo, F.; Sangiorgio, S.; Papa, G.; Adani, F.; Pilu, R. Brachytic2 mutation is able to counteract the main pleiotropic effects of brown midrib3 mutant in maize. Sci. Rep. 2022, 12, 2446. [Google Scholar] [CrossRef]
- Li, W.; Yan, J.; Zhang, Y.; Zhang, F.; Guan, Z.; Yao, Y.; Chang, Y.; Tu, H.; Li, X.; Wang, H.; et al. Serine protease NAL1 exerts pleiotropic functions through degradation of TOPLESS-related corepressor in rice. Nat. Plants 2023, 9, 1130–1142. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, W.; Liu, Y.; Yang, J.; Guo, X.; Zhang, J.; Pu, J.; Chen, N.; Zhang, W. Identification and Transcriptome Analysis of a Novel Allelic Mutant of NAL1 in Rice. Genes 2024, 15, 325. [Google Scholar] [CrossRef]
- Heisler, M.G.; Byrne, M.E. Progress in understanding the role of auxin in lateral organ development in plants. Curr. Opin. Plant Biol. 2020, 53, 73–79. [Google Scholar] [CrossRef]
- Kou, X.; Zhao, X.; Wu, B.; Wang, C.; Wu, C.; Yang, S.; Zhou, J.; Xue, Z. Auxin response factors are ubiquitous in plant growth and development, and involved in crosstalk between plant hormones: A review. Appl. Sci. 2022, 12, 1360. [Google Scholar] [CrossRef]
- Khan, M.N.; Nabi, G. Role of Auxin in vegetative growth, flowering, yield and fruit quality of Horticultural crops-A review. Pure Appl. Biol. 2023, 12, 1234–1241. [Google Scholar] [CrossRef]
- Zhang, Q.; Gong, M.; Xu, X.; Li, H.; Deng, W. Roles of auxin in the growth, development, and stress tolerance of horticultural plants. Cells 2022, 11, 2761. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhao, Y. A role for auxin in flower development. J. Integr. Plant Biol. 2007, 49, 99–104. [Google Scholar] [CrossRef]
- Yang, Z.; Xia, J.; Hong, J.; Zhang, C.; Wei, H.; Ying, W.; Sun, C.; Sun, L. Structural insights into auxin recognition and efflux by Arabidopsis PIN1. Nature 2022, 609, 611–615. [Google Scholar] [CrossRef]
- Vial-Pradel, S.; Keta, S.; Nomoto, M.; Luo, L.; Takahashi, H.; Suzuki, M.; Yokoyama, Y.; Sasabe, M.; Kojima, S.; Tada, Y.; et al. Arabidopsis zinc-finger-like protein ASYMMETRIC LEAVES2 (AS2) and two nucleolar proteins maintain gene body DNA methylation in the leaf polarity gene ETTIN (ARF3). Plant Cell Physiol. 2018, 59, 1385–1397. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Wu, M.F.; Winter, C.M.; Berns, M.C.; Nole-Wilson, S.; Yamaguchi, A.; Coupland, G.; Krizek, B.A.; Wagner, D. A molecular framework for auxin-mediated initiation of flower primordia. Dev. Cell 2013, 24, 271–282. [Google Scholar] [CrossRef]
- An, J.; Almasaud, R.A.; Bouzayen, M.; Zouine, M.; Chervin, C. Auxin and ethylene regulation of fruit set. Plant Sci. 2020, 292, 110381. [Google Scholar] [CrossRef]
- Li, B.J.; Bao, R.X.; Shi, Y.N.; Grierson, D.; Chen, K.S. Auxin response factors: Important keys for understanding regulatory mechanisms of fleshy fruit development and ripening. Hortic. Res. 2024, 11, uhae209. [Google Scholar] [CrossRef]
- Fenn, M.A.; Giovannoni, J.J. Phytohormones in fruit development and maturation. Plant J. 2021, 105, 446–458. [Google Scholar] [CrossRef]
- Sharif, R.; Su, L.; Chen, X.; Qi, X. Hormonal interactions underlying parthenocarpic fruit formation in horticultural crops. Hortic. Res. 2022, 9, uhab024. [Google Scholar] [CrossRef] [PubMed]
- Maupilé, L.; Chaib, J.; Boualem, A.; Bendahmane, A. Parthenocarpy, a pollination-independent fruit set mechanism to ensure yield stability. Trends Plant Sci. 2024, 29, 1254–1265. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Li, X.; Sun, T.P. Four class A AUXIN RESPONSE FACTORs promote tomato fruit growth despite suppressing fruit set. Nat. Plants 2023, 9, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Israeli, A.; Schubert, R.; Man, N.; Teboul, N.; Serrani Yarce, J.C.; Rosowski, E.E.; Wu, M.-F.; Levy, M.; Efroni, I.; Ljung, K.; et al. Modulating auxin response stabilizes tomato fruit set. Plant Physiol. 2023, 192, 2336–2355. [Google Scholar] [CrossRef]
- Li, Y.; Han, S.; Qi, Y. Advances in structure and function of auxin response factor in plants. J. Integr. Plant Biol. 2023, 65, 617–632. [Google Scholar] [CrossRef]
- Ezura, K.; Nomura, Y.; Ariizumi, T. Molecular, hormonal, and metabolic mechanisms of fruit set, the ovary-to-fruit transition, in horticultural crops. J. Exp. Bot. 2023, 74, 6254–6268. [Google Scholar] [CrossRef]
- Molesini, B.; Dusi, V.; Pennisi, F.; Pandolfini, T. How hormones and mads-box transcription factors are involved in controlling fruit set and parthenocarpy in tomato. Genes 2020, 11, 1441. [Google Scholar] [CrossRef]
- Hu, J.; Israeli, A.; Ori, N.; Sun, T.P. The interaction between DELLA and ARF/IAA mediates crosstalk between gibberellin and auxin signaling to control fruit initiation in tomato. Plant Cell 2018, 30, 1710–1728. [Google Scholar] [CrossRef]
- Figueiredo, D.D.; Köhler, C. Auxin: A molecular trigger of seed development. Genes Dev. 2018, 32, 479–490. [Google Scholar] [CrossRef]
- Cao, J.; Li, G.; Qu, D.; Li, X.; Wang, Y. Into the seed: Auxin controls seed development and grain yield. Int. J. Mol. Sci. 2020, 21, 1662. [Google Scholar] [CrossRef]
- Bernardi, J.; Li, Q.B.; Gao, Y.; Zhao, Y.; Battaglia, R.; Marocco, A.; Chourey, P.S. The auxin-deficient defective kernel18 (dek18) mutation alters the expression of seed-specific biosynthetic genes in maize. J. Plant Growth Regul. 2016, 35, 770–777. [Google Scholar] [CrossRef]
- de Folter, S.; Immink, R.G.; Kieffer, M.; Parenicova, L.; Henz, S.R.; Weigel, D.; Busscher, M.; Kooiker, M.; Colombo, L.; Kater, M.M.; et al. Comprehensive interaction map of the Arabidopsis MADS box transcription factors. Plant Cell 2005, 17, 1424–1433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mohanty, D.; Muzaffar, A.; Ni, M. Two MADS-box proteins, AGL9 and AGL15, recruit the FIS-PRC2 complex to trigger the phase transition from endosperm proliferation to embryo development in Arabidopsis. Mol. Plant 2024, 17, 1110–1128. [Google Scholar] [CrossRef]
- Terasaka, K.; Blakeslee, J.J.; Titapiwatanakun, B.; Peer, W.A.; Bandyopadhyay, A.; Makam, S.N.; Lee, O.R.; Richards, E.L.; Murphy, A.S.; Sato, F.; et al. PGP4, an ATP binding cassette P-glycoprotein, catalyzes auxin transport in Arabidopsis thaliana roots. Plant Cell 2005, 17, 2922–2939. [Google Scholar] [CrossRef]
- Zhu, F. Buckwheat proteins and peptides: Biological functions and food applications. Trends Food Sci. Technol. 2021, 110, 155–167. [Google Scholar] [CrossRef]
- Nishimura, M.; Ohkawara, T.; Sato, Y.; Satoh, H.; Suzuki, T.; Ishiguro, K.; Noda, T.; Morishita, T.; Nishihira, J. Effectiveness of rutin-rich Tartary buckwheat (Fagopyrum tataricum Gaertn.) “Manten-Kirari” in body weight reduction related to its antioxidant properties: A randomised, double-blind, placebo-controlled study. J. Funct. Foods 2016, 26, 460–469. [Google Scholar] [CrossRef]
- Zhang, L.; Li, X.; Ma, B.; Gao, Q.; Du, H.; Han, Y.; Li, Y.; Cao, Y.; Qi, M.; Zhu, Y.; et al. The tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance. Mol. Plant 2017, 10, 1224–1237. [Google Scholar] [CrossRef]
- Jin, J.; Ohanenye, I.C.; Udenigwe, C.C. Buckwheat proteins: Functionality, safety, bioactivity, and prospects as alternative plant-based proteins in the food industry. Crit. Rev. Food Sci. Nutr. 2022, 62, 1752–1764. [Google Scholar] [CrossRef]
- Logacheva, M.D.; Kasianov, A.S.; Vinogradov, D.V.; Samigullin, T.H.; Gelfand, M.S.; Makeev, V.J.; Penin, A.A. De novo sequencing and characterization of floral transcriptome in two species of buckwheat (Fagopyrum). BMC Genom. 2011, 12, 30. [Google Scholar] [CrossRef]
- Penin, A.A.; Kasianov, A.S.; Klepikova, A.V.; Kirov, I.V.; Gerasimov, E.S.; Fesenko, A.N.; Logacheva, M.D. High-resolution transcriptome atlas and improved genome assembly of common buckwheat, Fagopyrum esculentum. Front. Plant Sci. 2021, 12, 612382. [Google Scholar] [CrossRef]
- Wu, Q.; Bai, X.; Zhao, W.; Xiang, D.; Wan, Y.; Yan, J.; Zou, L.; Zhao, G. De novo assembly and analysis of tartary buckwheat (Fagopyrum tataricum Garetn.) transcriptome discloses key regulators involved in salt-stress response. Genes 2017, 8, 255. [Google Scholar] [CrossRef] [PubMed]
- Song, J.N.; Liu, X.H.; Wang, Y.Q.; Yang, H.B. Transcriptome analysis reveals salinity responses in four Tartary buckwheat cultivars. J. Plant Biochem. Biotechnol. 2021, 30, 564–578. [Google Scholar] [CrossRef]
- Yokosho, K.; Yamaji, N.; Ma, J.F. Global transcriptome analysis of Al-induced genes in an Al-accumulating species, common buckwheat (Fagopyrum esculentum Moench). Plant Cell Physiol. 2014, 55, 2077–2091. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, Q.; Rong, Y.; Tang, B.; Zhu, L.; Ren, R.; Shi, T.; Chen, Q. Transcriptome analysis revealed gene regulatory network involved in PEG-induced drought stress in Tartary buckwheat (Fagopyrum Tararicum). Peer J. 2021, 9, e11136. [Google Scholar] [CrossRef]
- Fang, Z.; Hou, Z.; Wang, S.; Liu, Z.; Wei, S.; Zhang, Y.; Song, J.; Yin, J. Transcriptome analysis reveals the accumulation mechanism of anthocyanins in buckwheat (Fagopyrum esculentum Moench) cotyledons and flowers. Int. J. Mol. Sci. 2019, 20, 1493. [Google Scholar] [CrossRef]
- Fang, X.; Zhang, Y.; Zhang, Y.; Huang, K.; Yang, W.; Li, X.; Zhang, Z.; Wu, K.; Xu, X.; Ruan, R.; et al. De novo transcriptome assembly and identification of genes related to seed size in common buckwheat (Fagopyrum esculentum M.). Breed. Sci. 2019, 69, 487–497. [Google Scholar] [CrossRef]
- Fang, X.; Wang, Y.; Cui, J.; Yue, L.; Jiang, A.; Liu, J.; Wu, Y.; He, X.; Li, C.; Zhang, J.; et al. Transcriptome and metabolome analyses reveal the key genes related to grain size of big grain mutant in Tartary Buckwheat (Fagopyrum tartaricum). Front. Plant Sci. 2022, 13, 1079212. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Hassan, M.J.; Li, Z.; Peng, Y. Indole-3-acetic acid improves drought tolerance of white clover via activating auxin, abscisic acid and jasmonic acid related genes and inhibiting senescence genes. BMC Plant Biol. 2020, 20, 1–12. [Google Scholar] [CrossRef]
- Liu, M.; Ma, Z.; Wang, A.; Zheng, T.; Huang, L.; Sun, W.; Zhang, Y.; Jin, W.; Zhan, J.; Cai, Y.; et al. Genome-wide investigation of the auxin response factor gene family in Tartary buckwheat (Fagopyrum tataricum). Int. J. Mol. Sci. 2018, 19, 3526. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, C.; Huang, S.; Chang, L.; Li, J.; Tang, H.; Dey, S.; Biswas, A.; Du, D.; Li, D.; et al. Key Cannabis Salt-Responsive Genes and Pathways Revealed by Comparative Transcriptome and Physiological Analyses of Contrasting Varieties. Agronomy 2021, 11, 2338. [Google Scholar] [CrossRef]
- Huo, D.; Hao, Y.; Zou, J.; Qin, L.; Wang, C.; Du, D. Integrated transcriptome and metabonomic analysis of key metabolic pathways in response to cadmium stress in novel buckwheat and cultivated species. Front. Plant Sci. 2023, 14, 1142814. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Jin, R.; Guo, J.; Zhang, F. Infection of embryonic callus with Agrobacterium enables high-speed transformation of maize. Int. J. Mol. Sci. 2019, 20, 279. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq: A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Bai, Y.C.; Li, C.L.; Zhang, J.W.; Li, S.J.; Luo, X.P.; Yao, H.P.; Chen, H.; Zhao, H.-X.; Park, S.-U.; Wu, Q. Characterization of two tartary buckwheat R2R3-MYB transcription factors and their regulation of proanthocyanidin biosynthesis. Physiol. Plant. 2014, 152, 431–440. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level the Deseq Package; European Molecular Biology Laboratory (EMBL): Heidelberg, Germany, 2012; Volume 10, p. f1000research. [Google Scholar]
- Zhang, Y.H.; Zeng, T.; Chen, L.; Huang, T.; Cai, Y.D. Determining protein–protein functional associations by functional rules based on gene ontology and KEGG pathway. BBA Proteins Proteom. 2021, 1869, 140621. [Google Scholar] [CrossRef]
- Yao, X.; Zhou, M.; Ruan, J.; He, A.; Ma, C.; Wu, W.; Lai, D.; Fan, Y.; Gao, A.; Weng, W.; et al. Genome-wide identification, evolution, and expression pattern analysis of the GATA gene family in tartary buckwheat (Fagopyrum tataricum). Int. J. Mol. Sci. 2022, 23, 12434. [Google Scholar] [CrossRef]
- Deng, K.; Yu, L.; Zheng, X.; Zhang, K.; Wang, W.; Dong, P.; Zhang, J.; Ren, M. Target of rapamycin is a key player for auxin signaling transduction in Arabidopsis. Front. Plant Sci. 2016, 7, 291. [Google Scholar] [CrossRef]
- De Smet, I.; Voß, U.; Lau, S.; Wilson, M.; Shao, N.; Timme, R.E.; Swarup, R.; Kerr, I.; Hodgman, C.; Bock, R.; et al. Unraveling the evolution of auxin signaling. Plant Physiol. 2011, 155, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.S.; Sharma, M.; Laxmi, A. Role of sugar and auxin crosstalk in plant growth and development. Physiol. Plant. 2022, 174, e13546. [Google Scholar] [CrossRef] [PubMed]
- Müllender, M.; Varrelmann, M.; Savenkov, E.I.; Liebe, S. Manipulation of auxin signalling by plant viruses. Mol. Plant Pathol. 2021, 22, 1449–1458. [Google Scholar] [CrossRef]
- Weijers, D.; Friml, J. SnapShot: Auxin signaling and transport. Cell 2009, 136, 1172. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hentrich, M.; Pollmann, S. Auxin-Oxylipin Crosstalk: Relationship of Antagonists F. J. Integr. Plant Biol. 2011, 53, 429–445. [Google Scholar] [CrossRef]
- Chen, Y.; Hao, X.; Cao, J. Small auxin upregulated RNA (SAUR) gene family in maize: Identification, evolution, and its phylogenetic comparison with Arabidopsis, rice, and sorghum. J. Integr. Plant Biol. 2014, 56, 133–150. [Google Scholar] [CrossRef]
- Stupar, R.M.; Springer, N.M. Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1 hybrid. Genetics 2006, 173, 2199–2210. [Google Scholar] [CrossRef]
- Jiao, Y.; Lori Tausta, S.; Gandotra, N.; Sun, N.; Liu, T.; Clay, N.K.; Ceserani, T.; Chen, M.; Ma, L.; Holford, M.; et al. A transcriptome atlas of rice cell types uncovers cellular, functional and developmental hierarchies. Nat. Genet. 2009, 41, 258–263. [Google Scholar] [CrossRef]
- Kagale, S.; Nixon, J.; Khedikar, Y.; Pasha, A.; Provart, N.J.; Clarke, W.E.; Bollina, V.; Robinson, S.J.; Coutu, C.; Dwayne, D.; et al. The developmental transcriptome atlas of the biofuel crop Camelina sativa. Plant J. 2016, 88, 879–894. [Google Scholar] [CrossRef]
- Armenta-Medina, A.; Gillmor, C.S.; Gao, P.; Mora-Macias, J.; Kochian, L.V.; Xiang, D.; Datla, R. Developmental and genomic architecture of plant embryogenesis: From model plant to crops. Plant Commun. 2021, 2, 100136. [Google Scholar] [CrossRef]
- Yang, F.; Lian, M.; Ma, H.; Feng, L.; Shen, X.; Chen, J.; Fang, J. Identification of key genes associated with papillary thyroid microcarcinoma characteristics by integrating transcriptome sequencing and weighted gene co-expression network analysis. Gene 2022, 811, 146086. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zenda, T.; Dong, A.; Yang, Y.; Wang, N.; Duan, H. Global transcriptome and weighted gene co-expression network analyses of growth-stage-specific drought stress responses in maize. Front. Genet. 2021, 12, 645443. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Sequence (5′-3′) |
|---|---|
| FtPinG0000387700 | AGCCGAGGAGGCACTTACTA//GGCTAGAGATCACAGACGGC |
| FtPinG0000683600 | AATGGTCTACCTGTGGCTGC//AATGCCTGAGGACTGACAGC |
| FtPinG0000764000 | CTCACCACAGGTTGGAGCAT//GCTGCAAGGACACCAATGTG |
| FtPinG0000807700 | AGCCGAAACGACACCTTCTT//GCTGCAAGGACACCAATGTG |
| FtPinG0000840000 | TAGGCGAAGACCCACCCATA//GCCTGATCTTTCAGGGTGCT |
| FtPinG0001741700 | GAGTGCTTGCAACTGCTTCC//CCGGTGCATCATCTCCTCAA |
| FtPinG0001971700 | AATGCGGACCAAGCAAACAC//TTCACTGCGAAGCTCATGGT |
| FtPinG0002469700 | GGAGTTGTGGCATGCTTGTG//TCATCAGTGTCTGGTTCCGC |
| FtPinG0002499600 | CCTCAAAGATCAGGCTGGCA//GCCTTGTGGATCCATCTGCT |
| FtPinG0002591700 | CCACAGGGACACATGGAACA//GACCTGCAACCTCCACATGA |
| FtPinG0003227500 | CGCAGGCATTTGCTGACTAC//TTGGGTTGCTCTTCGGACTC |
| FtPinG0003331700 | AAGCTGGCTCTGCTTCCATT//GCTCGTATCCGAAGCTGTCA |
| FtPinG0004636500 | CCTCCTCAGCTGATTTGCCA//TAATTGGTCGGTTGCCTGCT |
| FtPinG0005201700 | GGGCTGTTCAAAGCCAACAG//CCACCCGTTGAATCATCCCA |
| FtPinG0005575800 | CTCGTCCAAGCATGGGTTCT//AAGGCGAACGGAAACCAGAT |
| FtPinG0007231100 | TAATCGGCACAGCAGGGTTT//GTGACTCCCATTCCGCTTGA |
| FtPinG0008443000 | GATACGAGTACTCACGGCGG//CCCGCGGTATATGTGACGAA |
| FtPinG0008597700 | CGAAGATCCTCTGCCGTGTT//CAAAAAGAGTGCACGGTCGG |
| FtPinG0009506900 | TCACCATGGGAATTGGAGCC//TGGAGGCGGAAAATGGTCTC |
| Sample | Clean Reads Pairs | Clean Base (bp) | GC (%) | Q30 (%) | Total Read Pairs | Total Mapped Reads | Uniq Mapped Reads | Multiple Mapped Reads |
|---|---|---|---|---|---|---|---|---|
| Leaf-CK-1 | 20,728,710 | 6,218,613,000 | 46.32 | 89.90 | 20,728,710 | 19,559,891 (94.36%) | 19,092,767 (92.11%) | 467,124 (2.25%) |
| Leaf-CK-2 | 20,737,194 | 6,221,158,200 | 46.34 | 90.19 | 20,737,194 | 19,485,885 (93.97%) | 19,021,744 (91.73%) | 464,141 (2.24%) |
| Leaf-CK-3 | 20,117,528 | 6,035,258,400 | 46.09 | 90.46 | 20,117,528 | 18,874,534 (93.82%) | 18,412,786 (91.53%) | 461,748 (2.30%) |
| Leaf-1D-1 | 20,825,096 | 6,247,528,800 | 46.24 | 90.54 | 20,825,096 | 19,643,518 (94.33%) | 19,162,072 (92.01%) | 481,446 (2.31%) |
| Leaf-1D-2 | 21,184,145 | 6,355,243,500 | 46.17 | 89.97 | 21,184,145 | 19,936,268 (94.11%) | 19,395,678 (91.56%) | 540,590 (2.55%) |
| Leaf-1D-3 | 20,709,917 | 6,212,975,100 | 46.20 | 89.81 | 20,709,917 | 19,505,462 (94.18%) | 18,893,395 (91.23%) | 612,067 (2.96%) |
| Leaf-2D-1 | 20,764,665 | 6,229,399,500 | 46.49 | 90.09 | 20,764,665 | 19,408,654 (93.47%) | 18,763,601 (90.36%) | 645,053 (3.11%) |
| Leaf-2D-2 | 20,866,415 | 6,259,924,500 | 45.42 | 89.64 | 20,866,415 | 19,617,791 (94.02%) | 19,170,153 (91.87%) | 447,638 (2.15%) |
| Leaf-2D-3 | 21,089,542 | 6,326,862,600 | 46.06 | 90.26 | 21,089,542 | 19,691,229 (93.37%) | 19,066,009 (90.41%) | 625,220 (2.96%) |
| Leaf-3D-1 | 20,515,821 | 6,154,746,300 | 45.70 | 90.26 | 20,515,821 | 19,245,412 (93.81%) | 18,798,736 (91.63%) | 446,676 (2.18%) |
| Leaf-3D-2 | 20,706,805 | 6,212,041,500 | 46.03 | 90.19 | 20,706,805 | 19,426,273 (93.82%) | 18,913,196 (91.34%) | 513,077 (2.48%) |
| Leaf-3D-3 | 20,875,522 | 6,262,656,600 | 46.05 | 90.46 | 20,875,522 | 19,631,121 (94.04%) | 18,858,644 (90.34%) | 772,477 (3.70%) |
| Root-CK-1 | 20,444,463 | 6,168,821,000 | 44.48 | 90.12 | 20,444,463 | 19,145,496 (93.65%) | 18,751,891 (91.72%) | 393,605 (1.93%) |
| Root-CK-2 | 22,723,102 | 6,232,005,900 | 43.59 | 90.14 | 22,723,102 | 19,749,568 (86.91%) | 19,288,155 (84.88%) | 461,413 (2.03%) |
| Root-CK-3 | 22,117,521 | 6,361,707,400 | 44.49 | 90.08 | 22,117,521 | 19,376,695 (87.61%) | 18,863,241 (85.29%) | 513,454 (2.32%) |
| Root-1D-1 | 23,730,617 | 6,196,114,800 | 44.25 | 90.17 | 23,730,617 | 19,444,463 (81.93%) | 18,938,103 (79.80%) | 506,360 (2.13%) |
| Root-1D-2 | 19,731,775 | 6,374,886,000 | 44.10 | 90.15 | 19,731,775 | 19,723,102 (99.96%) | 19,325,198 (97.94%) | 397,904 (2.02%) |
| Root-1D-3 | 21,037,784 | 6,319,515,100 | 45.71 | 90.22 | 21,037,784 | 19,117,521 (90.87%) | 18,603,542 (88.43%) | 513,979 (2.44%) |
| Root-2D-1 | 23,131,324 | 6,220,229,600 | 45.27 | 90.22 | 23,131,324 | 19,730,617 (85.30%) | 19,247,665 (83.21%) | 482,952 (2.09%) |
| Root-2D-2 | 20,309,102 | 6,277,100,500 | 45.39 | 90.20 | 20,309,102 | 19,731,775 (97.16%) | 19,243,672 (94.75%) | 488,103 (2.40%) |
| Root-2D-3 | 20,975,996 | 6,206,517,800 | 46.16 | 90.23 | 20,975,996 | 19,037,784 (90.76%) | 18,694,823 (89.12%) | 342,961 (1.64%) |
| Root-3D-1 | 21,456,310 | 6,191,025,000 | 45.31 | 90.20 | 21,456,310 | 19,131,324 (89.16%) | 18,612,160 (86.74%) | 519,164 (2.42%) |
| Root-3D-2 | 20,059,779 | 6,098,242,500 | 45.24 | 90.15 | 20,059,779 | 19,309,102 (96.26%) | 19,025,500 (94.84%) | 283,602 (1.41%) |
| Root-3D-3 | 21,570,970 | 6,234,457,500 | 43.34 | 90.20 | 21,570,970 | 19,975,996 (92.61%) | 19,747,583 (91.55%) | 228,413 (1.06%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Lai, J.; Feng, C.; Li, L.; Zu, Q.; Li, J.; Du, D. Transcriptional Analysis of Tissues in Tartary Buckwheat Seedlings Under IAA Stimulation. Genes 2025, 16, 30. https://doi.org/10.3390/genes16010030
Gao Y, Lai J, Feng C, Li L, Zu Q, Li J, Du D. Transcriptional Analysis of Tissues in Tartary Buckwheat Seedlings Under IAA Stimulation. Genes. 2025; 16(1):30. https://doi.org/10.3390/genes16010030
Chicago/Turabian StyleGao, Yingying, Jialing Lai, Chenglu Feng, Luyang Li, Qihang Zu, Juan Li, and Dengxiang Du. 2025. "Transcriptional Analysis of Tissues in Tartary Buckwheat Seedlings Under IAA Stimulation" Genes 16, no. 1: 30. https://doi.org/10.3390/genes16010030
APA StyleGao, Y., Lai, J., Feng, C., Li, L., Zu, Q., Li, J., & Du, D. (2025). Transcriptional Analysis of Tissues in Tartary Buckwheat Seedlings Under IAA Stimulation. Genes, 16(1), 30. https://doi.org/10.3390/genes16010030