
Metabolomics and WGCNA Analyses Reveal the Underlying Mechanisms of Resistance to Botrytis cinerea in Hazelnut
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials, Growth Conditions, and B. cinerea Inoculation
2.2. Determination of the Physiological and Biochemical Parameters
2.3. Metabolomic Profile Detection and Analysis
2.4. Hub Gene Identification Using WGCNA
2.5. RNA Extraction and RT-qPCR Analysis
2.6. Statistics Analysis
3. Results
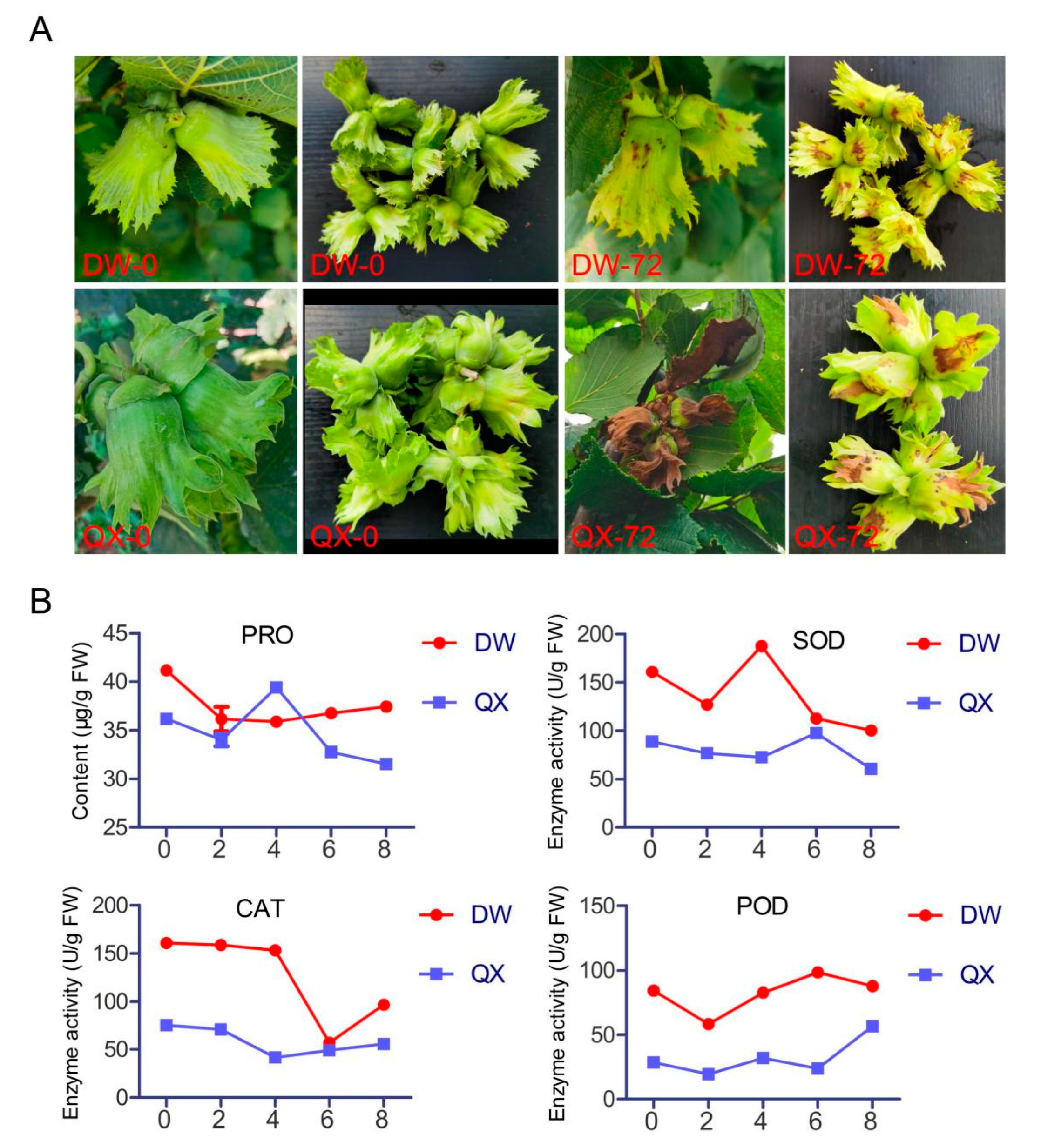
3.1. Phenotypic Characteristics and Physiological and Biochemical Parameters of QX and DW Following Inoculation with B. cinerea
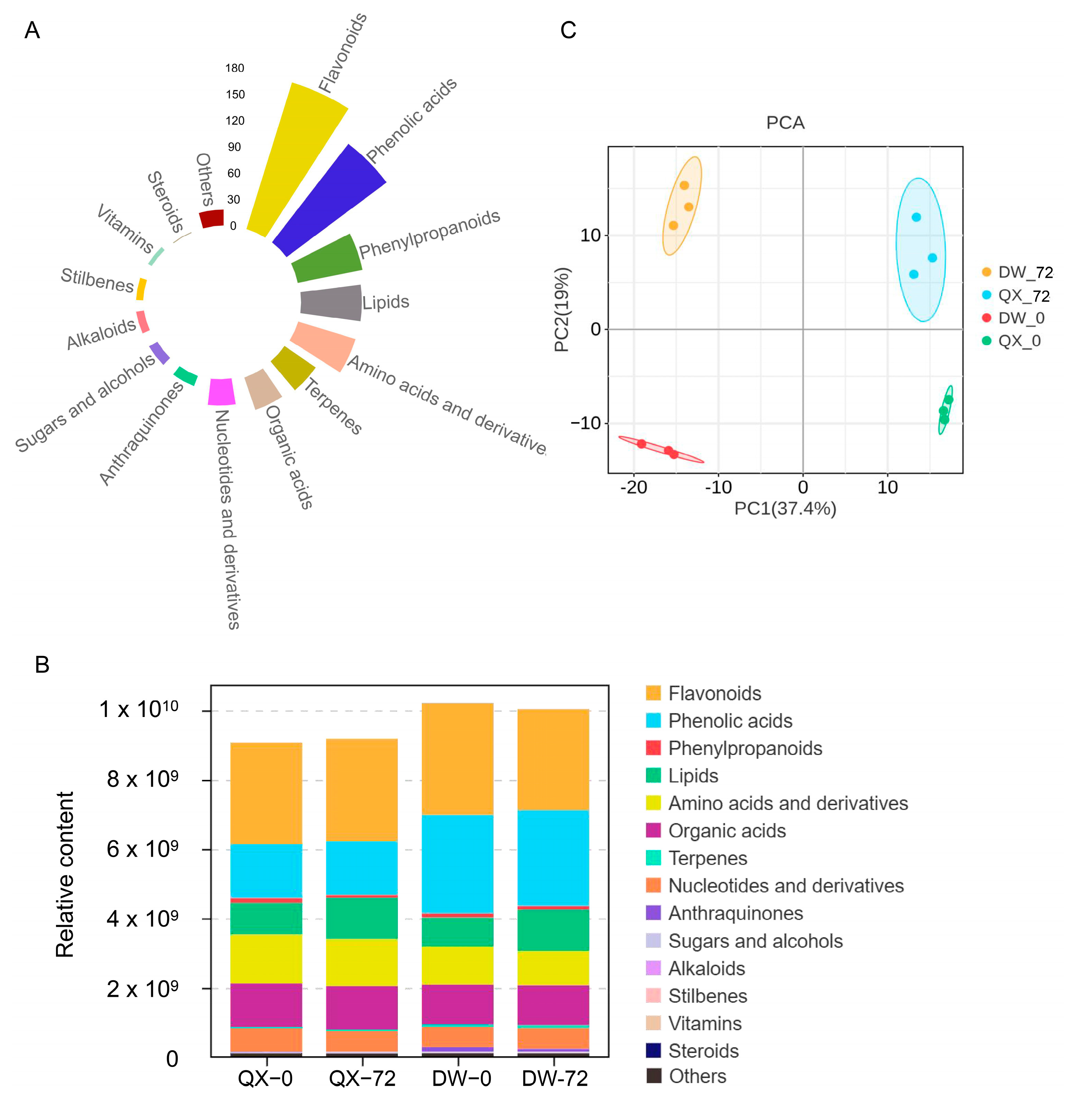
3.2. An Overview of Metabolomics
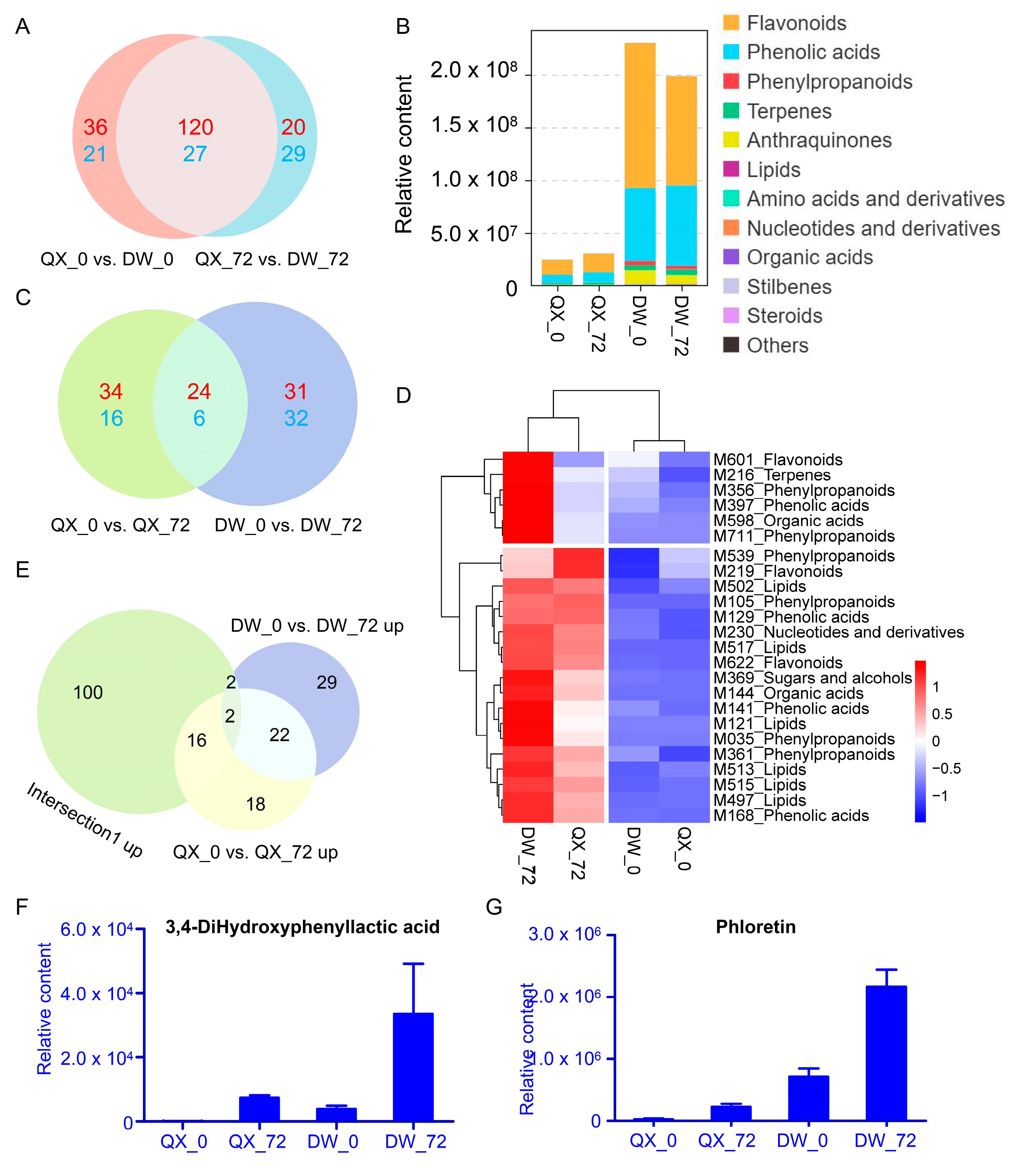
3.3. An Overview of DAMs
3.4. Venn Analysis of DAMs
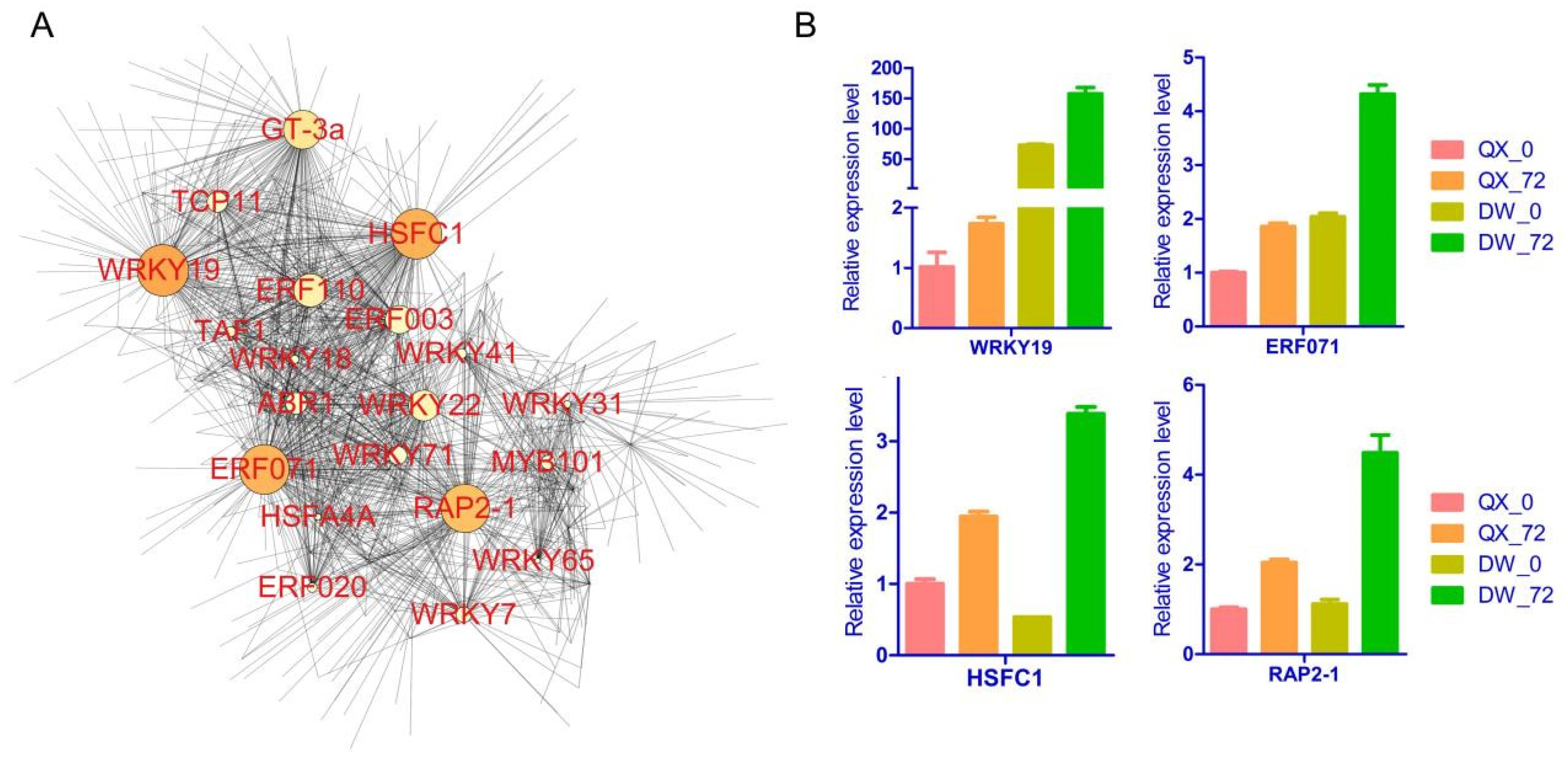
3.5. Gene Screening Using WGCNA
3.6. Network and RT-qPCR Analysis of Hub Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Silvestri, C.; Bacchetta, L.; Bellincontro, A.; Cristofori, V. Advances in cultivar choice, hazelnut orchard management, and nut storage to enhance product quality and safety: An overview. J. Sci. Food Agric. 2021, 101, 27–43. [Google Scholar] [CrossRef]
- Nicoletti, R.; Petriccione, M.; Curci, M.; Scortichini, M. Hazelnut-Associated bacteria and their Implications in crop management. Horticulturae 2022, 8, 1195. [Google Scholar] [CrossRef]
- Williamson, B.; Tudzynski, B.; Tudzynski, P.; van Kan, J.A. Botrytis cinerea: The cause of grey mould disease. Mol. Plant Pathol. 2007, 8, 561–580. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.; Van Kan, J.A.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed]
- van Kan, J.A. Licensed to kill: The lifestyle of a necrotrophic plant pathogen. Trends Plant Sci. 2006, 11, 247–253. [Google Scholar] [CrossRef] [PubMed]
- van Kan, J.A.; Shaw, M.W.; Grant-Downton, R.T. Botrytis species: Relentless necrotrophic thugs or endophytes gone rogue? Mol. Plant Pathol. 2014, 15, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M. The rising threat of fungicide resistance in plant pathogenic fungi: Botrytis as a case study. J. Chem. Biol. 2014, 7, 133–141. [Google Scholar] [CrossRef]
- Hahlbrock, K.; Bednarek, P.; Ciolkowski, I.; Hamberger, B.; Heise, A.; Liedgens, H.; Logemann, E.; Nürnberger, T.; Schmelzer, E.; Somssich, I.E.; et al. Non-self recognition, transcriptional reprogramming, and secondary metabolite accumulation during plant/pathogen interactions. Proc. Natl. Acad. Sci. USA 2003, 100, 14569–14576. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.Q.; Dong, X. Systemic acquired resistance: Turning local infection into global defense. Annu. Rev. Plant Biol. 2013, 64, 839–863. [Google Scholar] [CrossRef] [PubMed]
- Boller, T.; Felix, G. A renaissance of elicitors: Perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu. Rev. Plant Biol. 2009, 60, 379–406. [Google Scholar] [CrossRef]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Mengiste, T. Plant immunity to necrotrophs. Annu. Rev. Phytopathol. 2012, 50, 267–294. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Sun, P.; Li, Z.Y.; Zhang, F.J.; Wang, X.F.; You, C.X.; Zhang, C.L.; Zhang, Z. Plant disease resistance outputs regulated by AP2/ERF transcription factor family. Stress. Biol. 2024, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Liu, Y.; Huang, G.; Song, A.; Chen, S.; Jiang, J.; Chen, F.; Fang, W. Plant NAC transcription factors in the battle against pathogens. BMC Plant Biol. 2024, 24, 958. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, S.; Yu, Y.; Cui, N.; Yu, G.; Zhao, H.; Meng, X.; Fan, H. The pivotal role of MYB transcription factors in plant disease resistance. Planta 2023, 258, 16. [Google Scholar] [CrossRef] [PubMed]
- Javed, T.; Gao, S.J. WRKY transcription factors in plant defense. Trends Genet. 2023, 39, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.Y.; DeFalco, T.A.; Moeder, W.; Li, B.; Gong, Y.; Liu, X.M.; Taniguchi, M.; Lumba, S.; Toh, S.; Shan, L.; et al. Arabidopsis Ethylene Response Factor 8 (ERF8) has dual functions in ABA signaling and immunity. BMC Plant Biol. 2018, 18, 211. [Google Scholar] [CrossRef]
- Li, N.; Han, X.; Feng, D.; Yuan, D.; Huang, L.J. Signaling Crosstalk between salicylic acid and ethylene/jasmonate in plant defense: Do we understand what they are whispering? Int. J. Mol. Sci. 2019, 20, 671. [Google Scholar] [CrossRef] [PubMed]
- Clay, N.K.; Adio, A.M.; Denoux, C.; Jander, G.; Ausubel, F.M. Glucosinolate metabolites required for an Arabidopsis innate immune response. Science 2009, 323, 95–101. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y. Transcription factor VqERF114 regulates stilbene synthesis in Chinese wild Vitis quinquangularis by interacting with VqMYB35. Plant Cell Rep. 2019, 38, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Chen, Z.; Chen, R.; Shen, C. Environmental and genetic factors involved in plant protection-associated secondary metabolite biosynthesis pathways. Front. Plant Sci. 2022, 13, 877304. [Google Scholar] [CrossRef] [PubMed]
- Imano, S.; Fushimi, M.; Camagna, M.; Tsuyama-Koike, A.; Mori, H.; Ashida, A.; Tanaka, A.; Sato, I.; Chiba, S.; Kawakita, K.; et al. AP2/ERF transcription factor NbERF-IX-33 is involved in the regulation of phytoalexin production for the resistance of Nicotiana benthamiana to Phytophthora infestans. Front. Plant Sci. 2021, 12, 821574. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Li, X.; Hu, S.; Yang, D.; Abozeid, A.; Yang, Z.; Jiang, J.; Ren, Z.; Li, D.; Li, D.; et al. The critical role of phenylpropanoid biosynthesis pathway in Lily resistance against gray mold. Int. J. Mol. Sci. 2024, 25, 1068. [Google Scholar] [CrossRef] [PubMed]
- Schluttenhofer, C.; Yuan, L. Regulation of specialized metabolism by WRKY transcription factors. Plant Physiol. 2015, 167, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Liu, Q.; Wang, C.; Liang, J.; Liu, L.; Wang, Q. ZmWRKY79 positively regulates maize phytoalexin biosynthetic gene expression and is involved in stress response. J. Exp. Bot. 2018, 69, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Miao, H.; Chen, L.; Wang, M.; Xia, C.; Zeng, W.; Sun, B.; Zhang, F.; Zhang, S.; Li, C.; et al. WRKY33-mediated indolic glucosinolate metabolic pathway confers resistance against Alternaria brassicicola in Arabidopsis and Brassica crops. J. Integr. Plant Biol. 2022, 64, 1007–1019. [Google Scholar] [CrossRef]
- Soni, N.; Altartouri, B.; Hegde, N.; Duggavathi, R.; Nazarian-Firouzabadi, F.; Kushalappa, A.C. TaNAC032 transcription factor regulates lignin-biosynthetic genes to combat Fusarium head blight in wheat. Plant Sci. 2021, 304, 110820. [Google Scholar] [CrossRef] [PubMed]
- Frerigmann, H.; Gigolashvili, T. Update on the role of R2R3-MYBs in the regulation of glucosinolates upon sulfur deficiency. Front. Plant Sci. 2014, 5, 626. [Google Scholar] [CrossRef] [PubMed]
- Baskar, V.; Park, S.W. Molecular characterization of BrMYB28 and BrMYB29 paralogous transcription factors involved in the regulation of aliphatic glucosinolate profiles in Brassica rapa ssp. pekinensis. Comptes Rendus Biol. 2015, 338, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.J.; Dong, D.F.; Wang, G.X.; Dong, F.X.; Liang, L.S.; Ma, Q.H. Progresses on the hazelnut cross breeding of Corylus heterophylla fisch × Corylus avellana L. in China. Acta Hortic. 2012, 940, 233–238. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, X.; Zheng, J.; Liu, G.; Chen, L. Importance of cell wall permeability and cell wall degrading enzymes during infection of Botrytis cinerea in hazelnut. Forests 2023, 14, 565. [Google Scholar] [CrossRef]
- Kanehisa, M. KEGG Bioinformatics resource for plant genomics and metabolomics. Methods Mol. Biol. 2016, 1374, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, Y.; Zheng, Z.; Hao, J.; Chen, L. Comparative transcriptomic analysis reveals the molecular responses in two contrasting hazelnut varieties against Botrytis cinerea Infection. Forests 2023, 14, 493. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hou, S.; Zhao, T.; Yang, D.; Li, Q.; Liang, L.; Wang, G.; Ma, Q. Selection and validation of reference genes for quantitative RT-PCR analysis in Corylus heterophylla Fisch. × Corylus avellana L. Plants 2021, 10, 159. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.; Chen, J.; Huang, W.; Huang, G.; Deng, M.; Hong, S.; Ai, P.; Gao, C.; Zhou, H. OmicShare tools: A zero-code interactive online platform for biological data analysis and visualization. iMeta 2024, 3, e228. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A.; Mundt, F. factoextra: Extract and Visualize the Results of Multivariate Data Analyses. 2017, 76. Available online: https://cran.r-project.org/web/packages/factoextra/readme/README.html (accessed on 3 August 2024).
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.B.; Wada, S.; Stockwell, V.O.; Wiman, N.G. Susceptibility of some Corylus avellana L. Cultivars to Xanthomonas arboricola pv. corylina. Front. Plant Sci. 2021, 12, 800339. [Google Scholar] [CrossRef] [PubMed]
- Pacchiarelli, A.; Silvestri, C.; Cristofori, V. Advances in sucker control for sustainable European hazelnut (Corylus avellana L.) cultivation. Plants 2022, 11, 3416. [Google Scholar] [CrossRef] [PubMed]
- Hayat, S.; Hayat, Q.; Alyemeni, M.N.; Wani, A.S.; Pichtel, J.; Ahmad, A. Role of proline under changing environments: A review. Plant Signal. Behav. 2012, 7, 1456–1466. [Google Scholar] [CrossRef]
- Sewelam, N.; Kazan, K.; Schenk, P.M. Global Plant stress signaling: Reactive oxygen species at the cross-road. Front. Plant Sci. 2016, 7, 187. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.; Sathe, S.K. Chemical composition of selected edible nut seeds. J. Agric. Food Chem. 2006, 54, 4705–4714. [Google Scholar] [CrossRef] [PubMed]
- Bottone, A.; Cerulli, A.; DʼUrso, G.; Masullo, M.; Montoro, P.; Napolitano, A.; Piacente, S. Plant specialized metabolites in hazelnut (Corylus avellana) kernel and byproducts: An update on chemistry, biological activity, and analytical aspects. Planta Med. 2019, 85, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.P.; Ferreira, I.C.; Marcelino, F.; Valentão, P.; Andrade, P.B.; Seabra, R.; Estevinho, L.; Bento, A.; Pereira, J.A. Phenolic compounds and antimicrobial activity of olive (Olea europaea L. Cv. Cobrançosa) leaves. Molecules 2007, 12, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Proestos, C.; Chorianopoulos, N.; Nychas, G.J.; Komaitis, M. RP-HPLC analysis of the phenolic compounds of plant extracts. investigation of their antioxidant capacity and antimicrobial activity. J. Agric. Food Chem. 2005, 53, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.P.; Singh, R.K.; Jaiswal, H.K.; Kumar, V.; Maurya, S. Rhizobium-mediated induction of phenolics and plant growth promotion in rice (Oryza sativa L.). Curr. Microbiol. 2006, 52, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Perez de Souza, L.; Benina, M.; Fernie, A.R. The style and substance of plant flavonoid decoration; towards defining both structure and function. Phytochemistry 2020, 174, 112347. [Google Scholar] [CrossRef] [PubMed]
- Haipeng, Z.; Xiangyu, D.; Xiaomeng, C. Plant immune inducer ZNC promotes rutin accumulation and enhances resistance to Botrytis cinerea in tomato. Stress. Biol. 2023, 3, 36. [Google Scholar] [CrossRef]
- Lijun, W.; Dezheng, G.; Guangdong, Z. Group IIc WRKY transcription factors regulate cotton resistance to Fusarium oxysporum by promoting GhMKK2-mediated flavonoid biosynthesis. New Phytol. 2022, 236, 249–265. [Google Scholar] [CrossRef]
- Lahari, Z.; van Boerdonk, S.; Omoboye, O.O.; Reichelt, M.; Höfte, M.; Gershenzon, J.; Gheysen, G.; Ullah, C. Strigolactone deficiency induces jasmonate, sugar and flavonoid phytoalexin accumulation enhancing rice defense against the blast fungus Pyricularia oryzae. New Phytol. 2024, 241, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Hu, L.; Li, Y.; Chen, X.; Zhang, Z.; Liu, B.; Li, P.; Gong, X.; Ma, F. MdUGT88F1-mediated phloridzin biosynthesis regulates apple development and valsa canker resistance. Plant Physiol. 2019, 180, 2290–2305. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wu, P.; Yang, L.; Liu, C.; Guo, P.; Wang, H.; Wang, S.; Xu, F.; Zhuang, Q.; Tong, X.; et al. Transcriptomics and metabolomics reveal the induction of flavonoid biosynthesis pathway in the interaction of Stylosanthes-Colletotrichum gloeosporioides. Genomics 2021, 113, 2702–2716. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, H.; Yuan, Y. A Comparison of the flavonoid biosynthesis mechanisms of dendrobium species by analyzing the transcriptome and metabolome. Int. J. Mol. Sci. 2022, 23, 1980. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Huang, H.; Tan, C.; Gao, L.; Wan, S.; Zhu, B.; Chen, D.; Zhu, B. Transcriptome and WGCNA analyses reveal key genes regulating anthocyanin biosynthesis in purple sprout of pak choi (Brassica rapa L. ssp. chinensis). Int. J. Mol. Sci. 2024, 25, 1736. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhu, C.; Xu, C.; Sun, J.; Grierson, D.; Zhang, B.; Chen, K. Integration of metabolite profiling and transcriptome analysis reveals genes related to volatile terpenoid metabolism in finger citron (C. medica var. sarcodactylis). Molecules 2019, 24, 2564. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.; Lu, L.; Liu, J.; Cui, Y.; Liu, H.; Zhang, Y.; Zheng, Z.; Yang, W. Metabolomics and WGCNA Analyses Reveal the Underlying Mechanisms of Resistance to Botrytis cinerea in Hazelnut. Genes 2025, 16, 2. https://doi.org/10.3390/genes16010002
Sun J, Lu L, Liu J, Cui Y, Liu H, Zhang Y, Zheng Z, Yang W. Metabolomics and WGCNA Analyses Reveal the Underlying Mechanisms of Resistance to Botrytis cinerea in Hazelnut. Genes. 2025; 16(1):2. https://doi.org/10.3390/genes16010002
Chicago/Turabian StyleSun, Jun, Liyuan Lu, Juanjuan Liu, Yanhong Cui, Hanqi Liu, Yue Zhang, Zeyang Zheng, and Weicong Yang. 2025. "Metabolomics and WGCNA Analyses Reveal the Underlying Mechanisms of Resistance to Botrytis cinerea in Hazelnut" Genes 16, no. 1: 2. https://doi.org/10.3390/genes16010002
APA StyleSun, J., Lu, L., Liu, J., Cui, Y., Liu, H., Zhang, Y., Zheng, Z., & Yang, W. (2025). Metabolomics and WGCNA Analyses Reveal the Underlying Mechanisms of Resistance to Botrytis cinerea in Hazelnut. Genes, 16(1), 2. https://doi.org/10.3390/genes16010002