
Biallelic NDUFA4 Deletion Causes Mitochondrial Complex IV Deficiency in a Patient with Leigh Syndrome

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
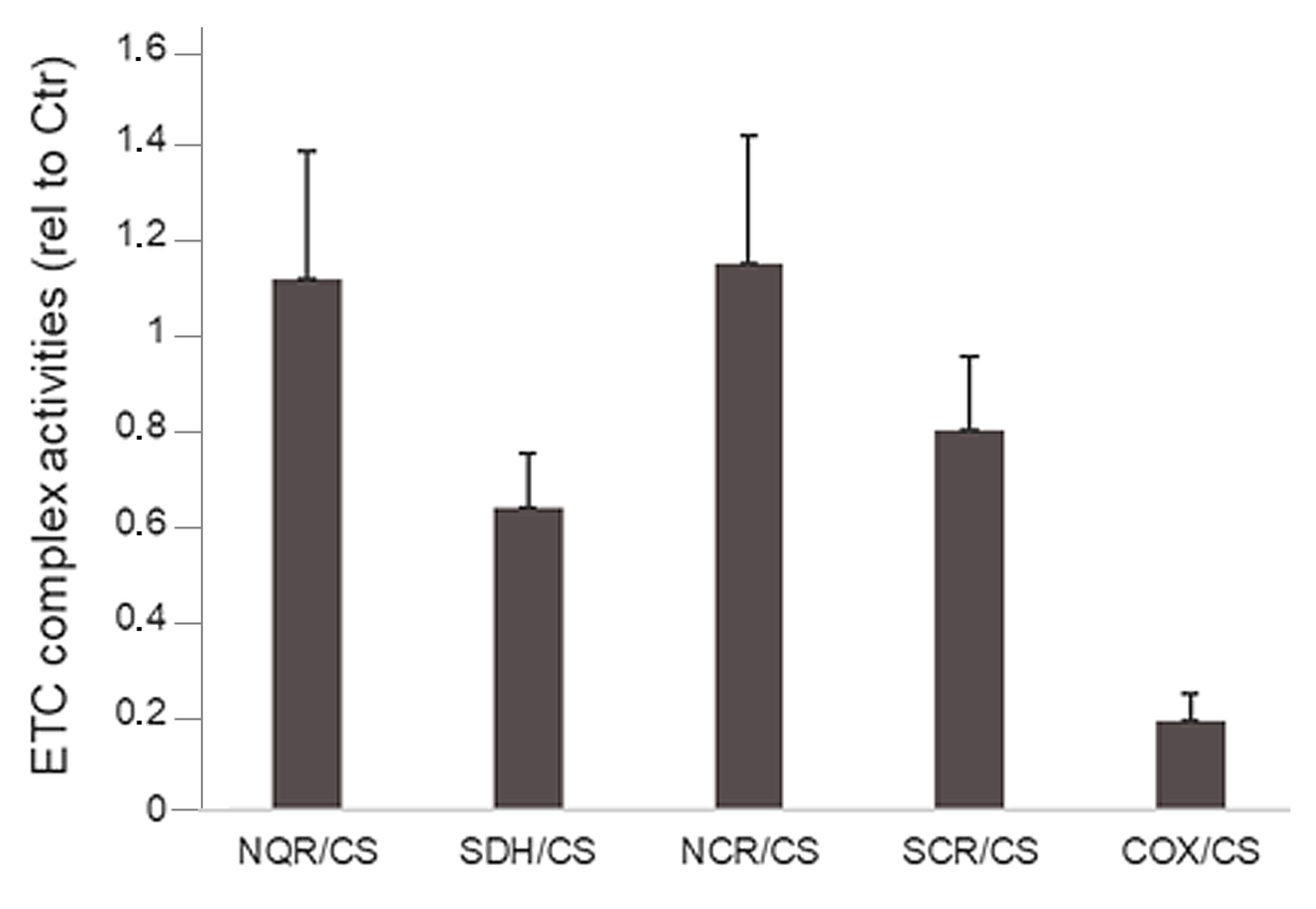
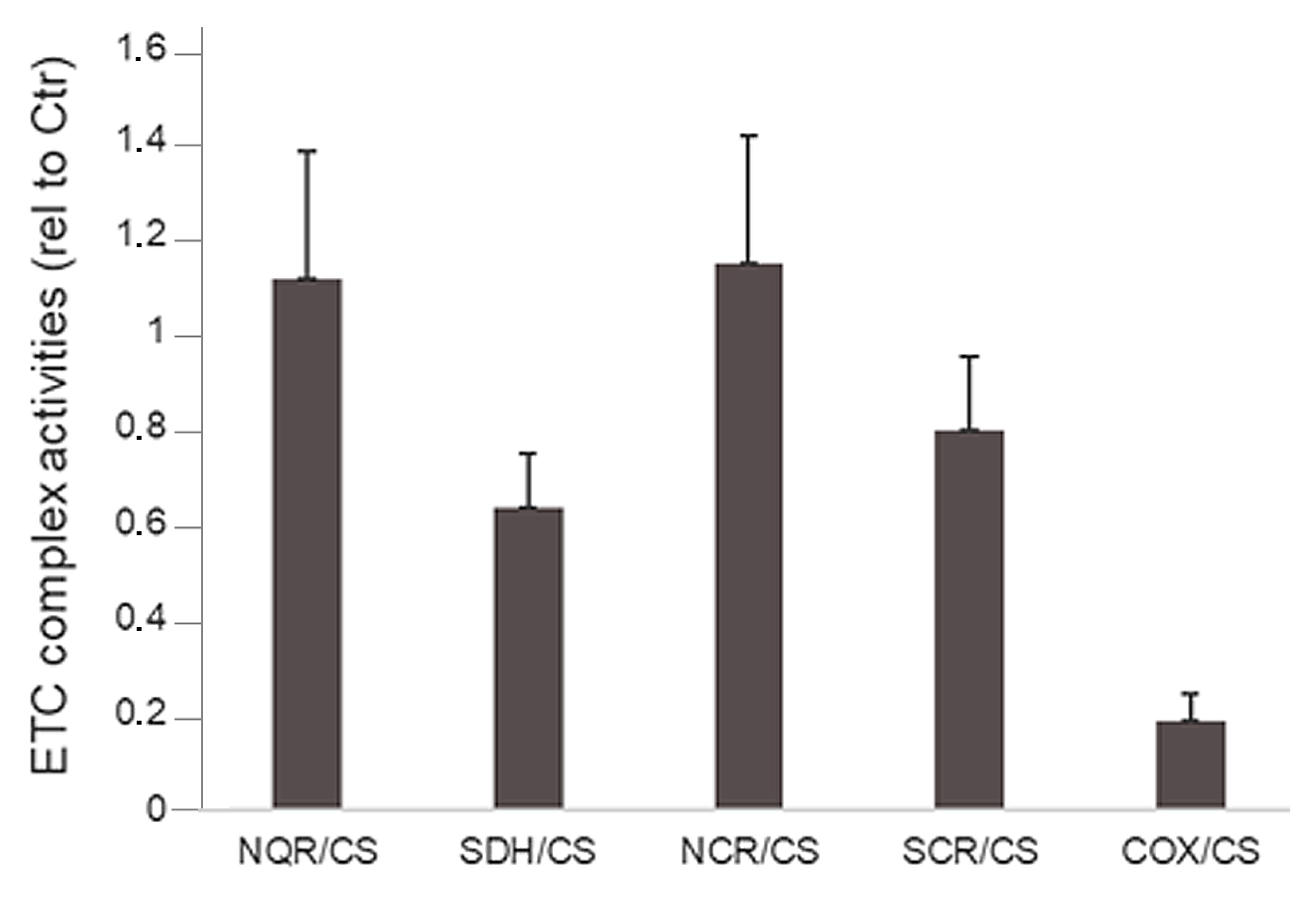
3. Results
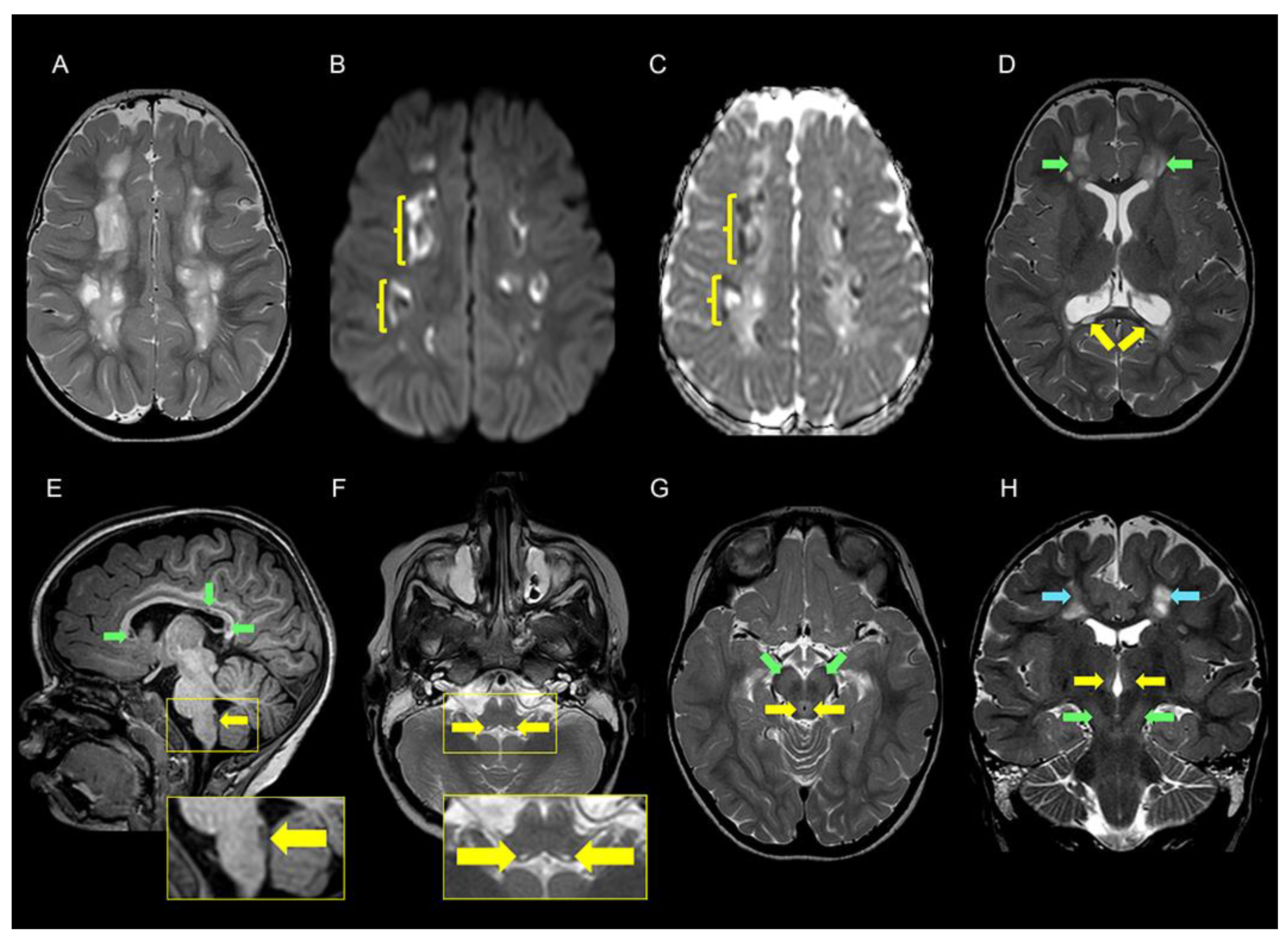
3.1. Clinical Description
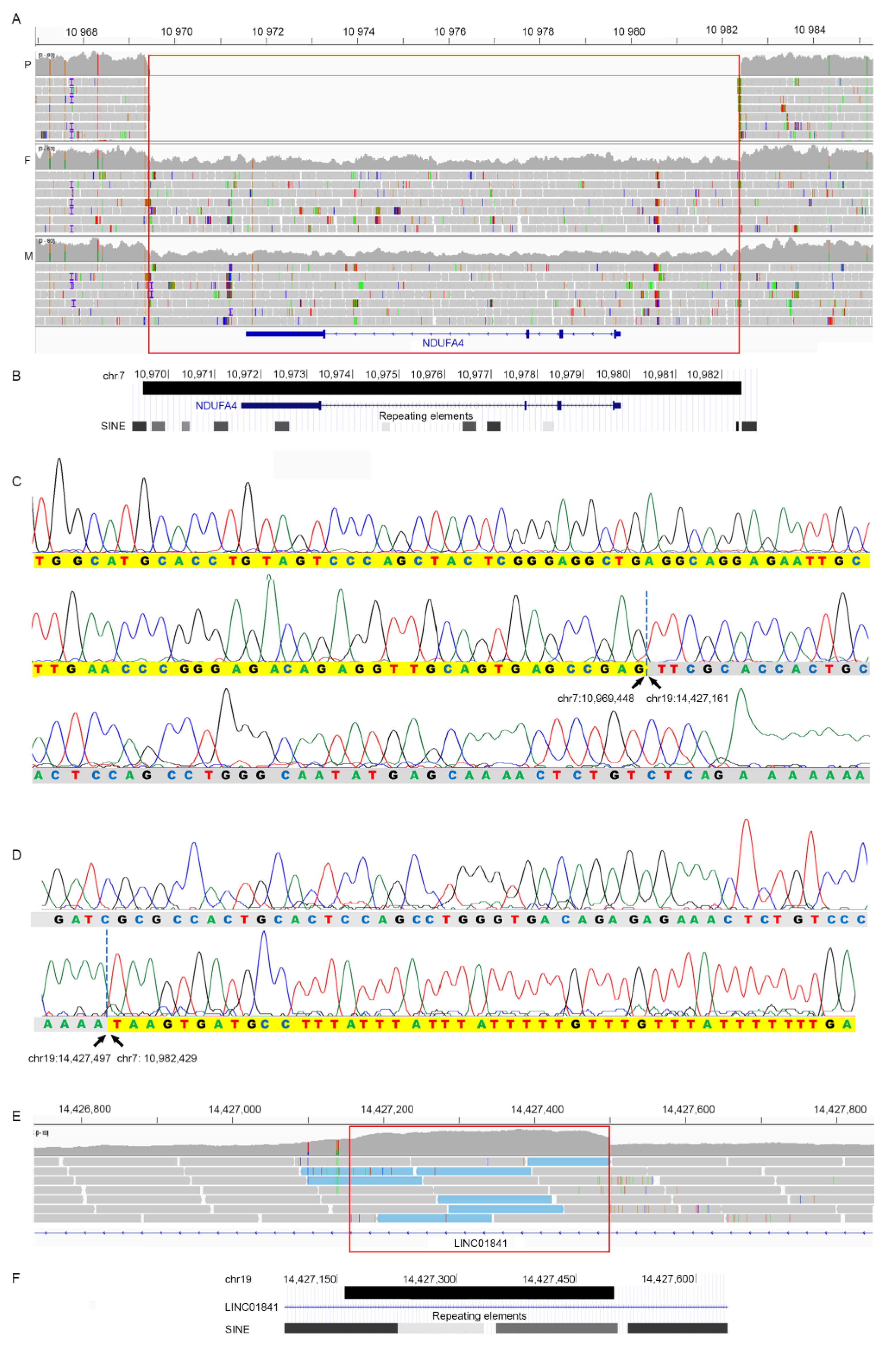
3.2. Genetic Findings
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahman, S. Mitochondrial disease in children. J. Intern. Med. 2020, 287, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Skladal, D.; Halliday, J.; Thorburn, D.R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 2003, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F. Primary Mitochondrial Disorders Overview. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Stromme, P.; Magnus, P.; Kanavin, O.J.; Rootwelt, T.; Woldseth, B.; Abdelnoor, M. Mortality in childhood progressive encephalopathy from 1985 to 2004 in Oslo, Norway: A population-based study. Acta Paediatr. 2008, 97, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Schlieben, L.D.; Prokisch, H. The Dimensions of Primary Mitochondrial Disorders. Front. Cell Dev. Biol. 2020, 8, 600079. [Google Scholar] [CrossRef] [PubMed]
- Dennerlein, S.; Rehling, P.; Richter-Dennerlein, R. Cytochrome c oxidase biogenesis-from translation to early assembly of the core subunit COX1. FEBS Lett. 2023, 597, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landazuri, M.O.; Enriquez, J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012, 16, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.; Rahman, S.; Wedatilake, Y.; Polke, J.M.; Cirak, S.; Foley, A.R.; Sailer, A.; Hurles, M.E.; Stalker, J.; Hargreaves, I.; et al. NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013, 3, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.S.; Taanman, J.W. NDUFA4 (Renamed COXFA4) Is a Cytochrome-c Oxidase Subunit. Trends Endocrinol. Metab. 2018, 29, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Wibom, R.; Hagenfeldt, L.; von Dobeln, U. Measurement of ATP production and respiratory chain enzyme activities in mitochondria isolated from small muscle biopsy samples. Anal. Biochem. 2002, 311, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Cho, C.S.; Han, K.; Lee, J. Structural Variation of Alu Element and Human Disease. Genom. Inform. 2016, 14, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordonez, A.; Corral-Escariz, M.; Soro, I.; Lopez-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Clayton, S.A.; Daley, K.K.; MacDonald, L.; Fernandez-Vizarra, E.; Bottegoni, G.; O’Neil, J.D.; Major, T.; Griffin, D.; Zhuang, Q.; Adewoye, A.B.; et al. Inflammation causes remodeling of mitochondrial cytochrome c oxidase mediated by the bifunctional gene C15orf48. Sci. Adv. 2021, 7, eabl5182. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Chen, C.; Du, K.; Li, L.S.; Li, R.; Lei, T.Y.; Deng, Q.; Wang, D.; Yu, Q.X.; Yang, X.; et al. Ndufa4 Regulates the Proliferation and Apoptosis of Neurons via miR-145a-5p/Homer1/Ccnd2. Mol. Neurobiol. 2023, 60, 2986–3003. [Google Scholar] [CrossRef] [PubMed]
- Brischigliaro, M.; Zeviani, M. Cytochrome c oxidase deficiency. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148335. [Google Scholar] [CrossRef] [PubMed]
- McCormick, E.M.; Keller, K.; Taylor, J.P.; Coffey, A.J.; Shen, L.; Krotoski, D.; Harding, B.; NICHD ClinGen U24 Mitochondrial Disease Gene Curation Expert Panel; Gai, X.; Falk, M.J.; et al. Expert Panel Curation of 113 Primary Mitochondrial Disease Genes for the Leigh Syndrome Spectrum. Ann. Neurol. 2023, 94, 696–712. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Patients | Patient 1 | Four Patients Reported in the Literature [8] |
|---|---|---|
| Age at last examination; gender | 4 y; female | 2 females (II-3 32 y; II-6 26 y, died), 2 males (II-4 34 y; II-13 8 y, died) |
| Country of origin | Somalia | Pakistan |
| Perinatal history | Normal | Low birth weight (2/4), poor growth (2/4), resuscitation (1/4) |
| Neurological functioning | ||
| Cognition | Language delay, learning difficulties | Language delay, learning difficulties (4/4) |
| Motor skills | Delayed; regression followed by slight improvement | Delayed (3/4); regression from age 4 y (1/4) |
| Pyramidal tract signs | Hyperreflexia, spasticity | Not reported |
| Extrapyramidal signs | Dystonic positioning of the feet | Dystonia (3/4) |
| Cerebellar signs | Ataxia, intention- and action tremor, broad-based gait | Ataxia (2/4), nystagmus (1/4) |
| Lactate measurements | ||
| CSF | Elevated 1 | Not reported |
| Plasma | Elevated 2 | Congenital lactic acidosis (4/4) |
| Cerebral MRI | ||
| Cerebral hemispheres | Leukoencephalopathy, widespread with multiple cavities 3 | Scattered signal changes in deep white matter |
| Corpus callosum | Atrophy, multiple cysts 4 | Not reported |
| Brain stem and mesencephalon | Signal changes in pons and medulla oblongata 5 and periaqueductal grey matter 6 | Signal changes in pons and medulla oblongata |
| Basal ganglia | Signal changes in substantia nigra and thalami 7 | Signal changes in medial thalami |
| Cerebellum | Normal | Signal changes in cerebellum and right middle cerebellar peduncle |
| Diagnosis | Leigh syndrome | Leigh syndrome (4/4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Misceo, D.; Strømme, P.; Bitarafan, F.; Chawla, M.S.; Sheng, Y.; Bach de Courtade, S.M.; Eide, L.; Frengen, E. Biallelic NDUFA4 Deletion Causes Mitochondrial Complex IV Deficiency in a Patient with Leigh Syndrome. Genes 2024, 15, 500. https://doi.org/10.3390/genes15040500
Misceo D, Strømme P, Bitarafan F, Chawla MS, Sheng Y, Bach de Courtade SM, Eide L, Frengen E. Biallelic NDUFA4 Deletion Causes Mitochondrial Complex IV Deficiency in a Patient with Leigh Syndrome. Genes. 2024; 15(4):500. https://doi.org/10.3390/genes15040500
Chicago/Turabian StyleMisceo, Doriana, Petter Strømme, Fatemeh Bitarafan, Maninder Singh Chawla, Ying Sheng, Sandra Monica Bach de Courtade, Lars Eide, and Eirik Frengen. 2024. "Biallelic NDUFA4 Deletion Causes Mitochondrial Complex IV Deficiency in a Patient with Leigh Syndrome" Genes 15, no. 4: 500. https://doi.org/10.3390/genes15040500
APA StyleMisceo, D., Strømme, P., Bitarafan, F., Chawla, M. S., Sheng, Y., Bach de Courtade, S. M., Eide, L., & Frengen, E. (2024). Biallelic NDUFA4 Deletion Causes Mitochondrial Complex IV Deficiency in a Patient with Leigh Syndrome. Genes, 15(4), 500. https://doi.org/10.3390/genes15040500