Intraspecific and Intrageneric Genomic Variation across Three Sedum Species (Crassulaceae): A Plastomic Perspective

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Plastome Sequencing
2.2. Plastome Assembly, Annotation, and Nucleotide Compositions Analysis
2.3. Analyses of Dispersed and Simple Sequence Repeats
2.4. Codon Usage Analysis of the Plastome
2.5. Phylogenetic Analysis
3. Results
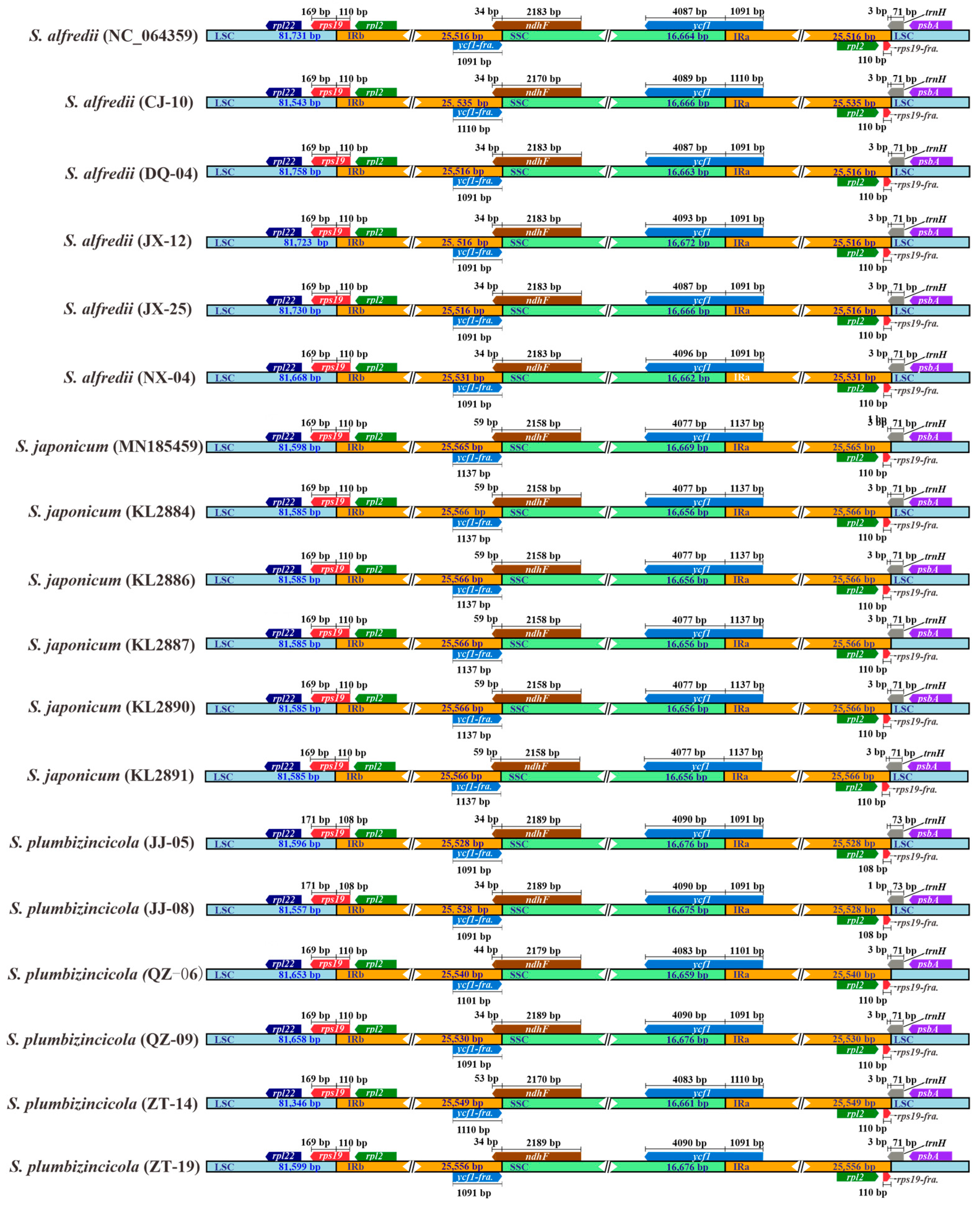
3.1. Comparative Analysis of Composition and Structure
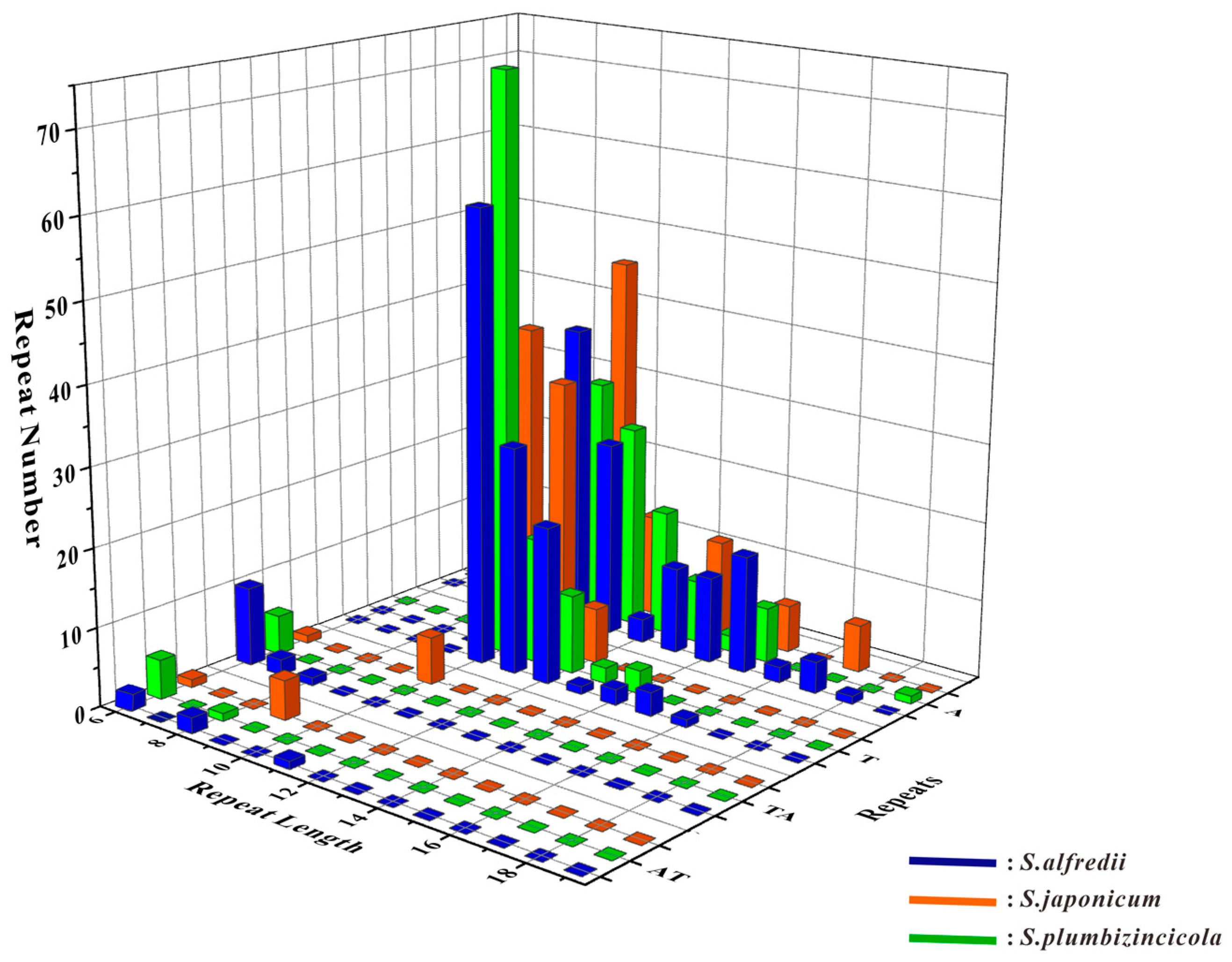
3.2. Analysis of Repeats in Three Sedum Species
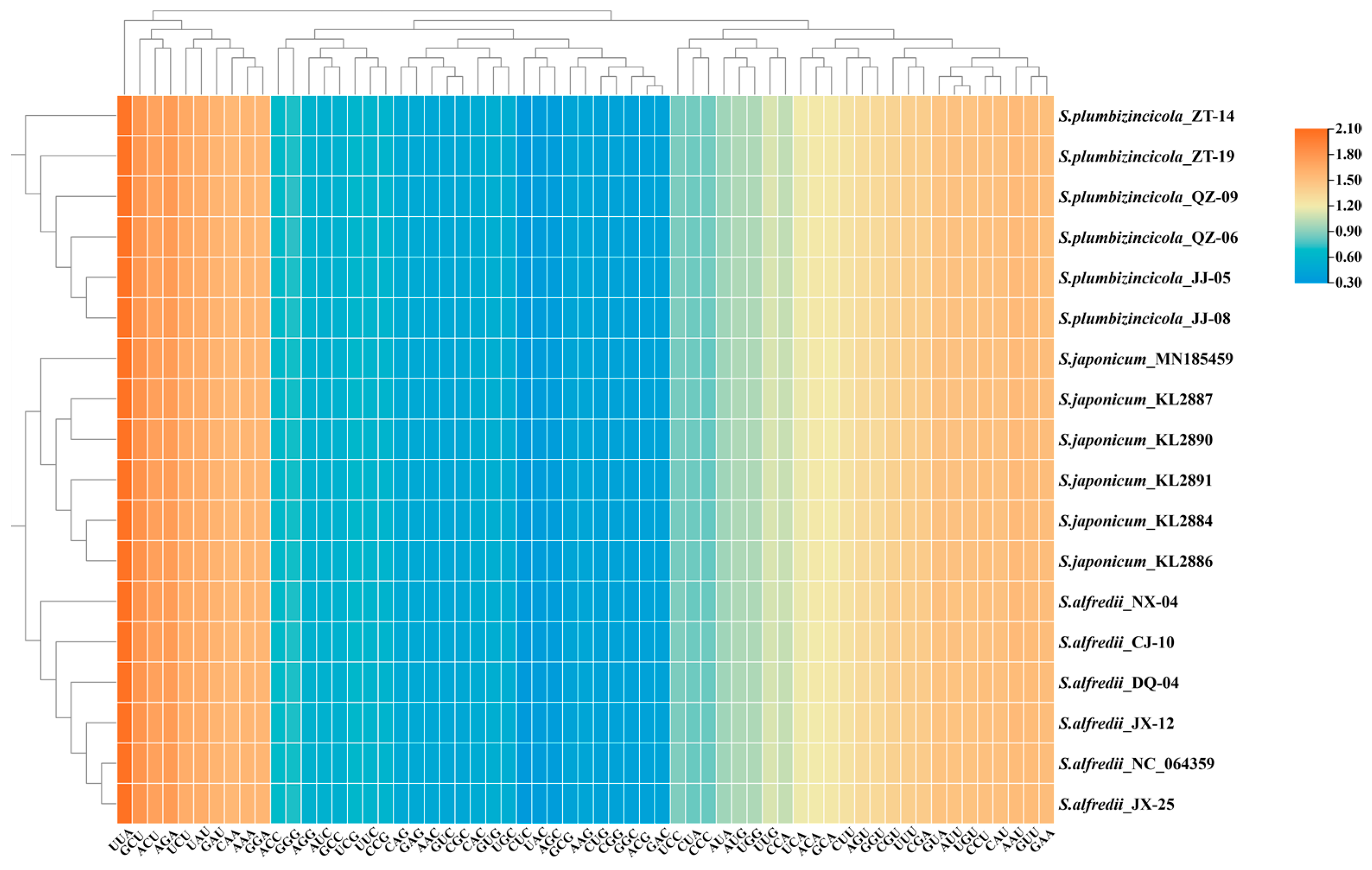
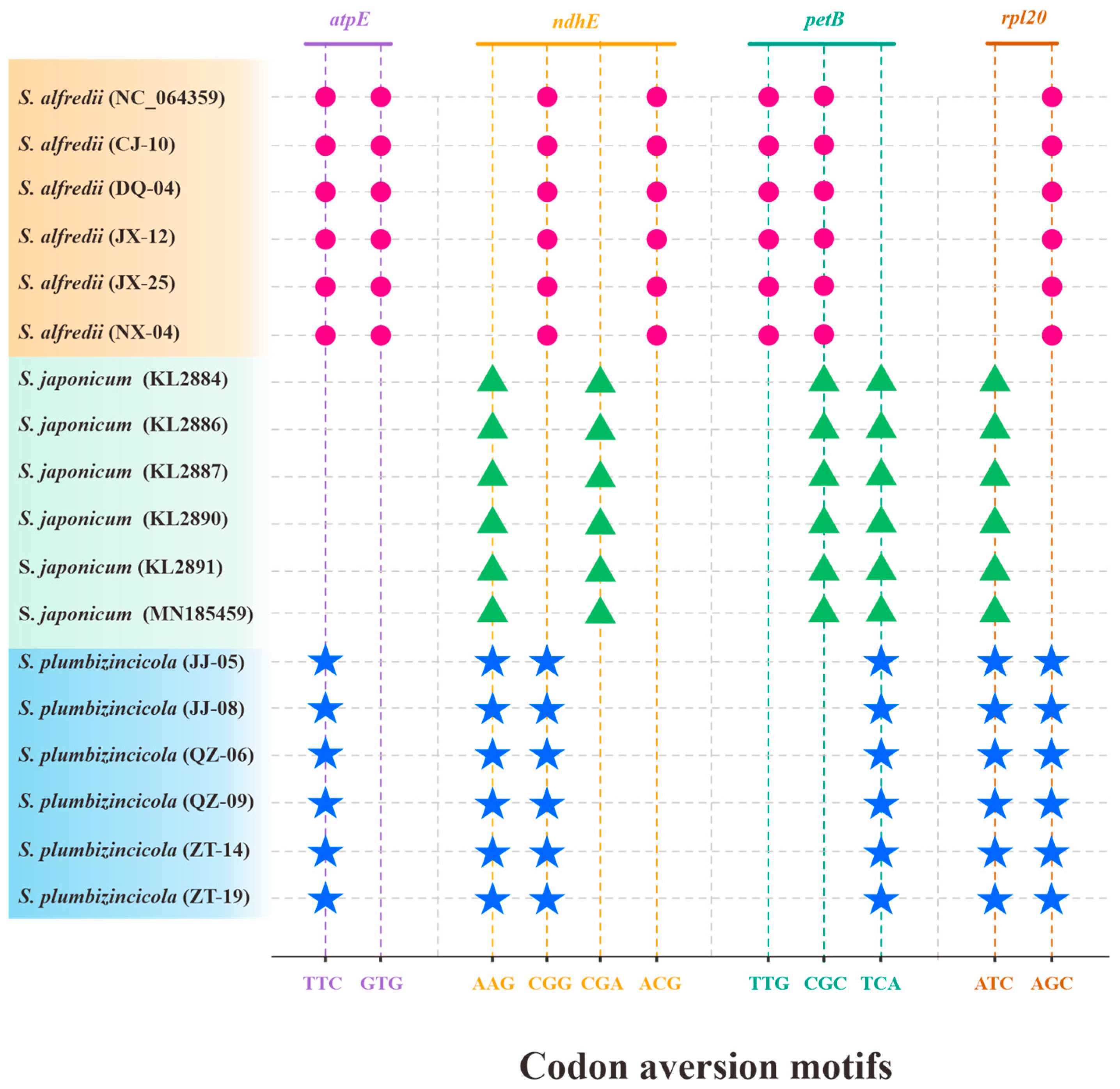
3.3. Codon Usage and Aversion Patterns
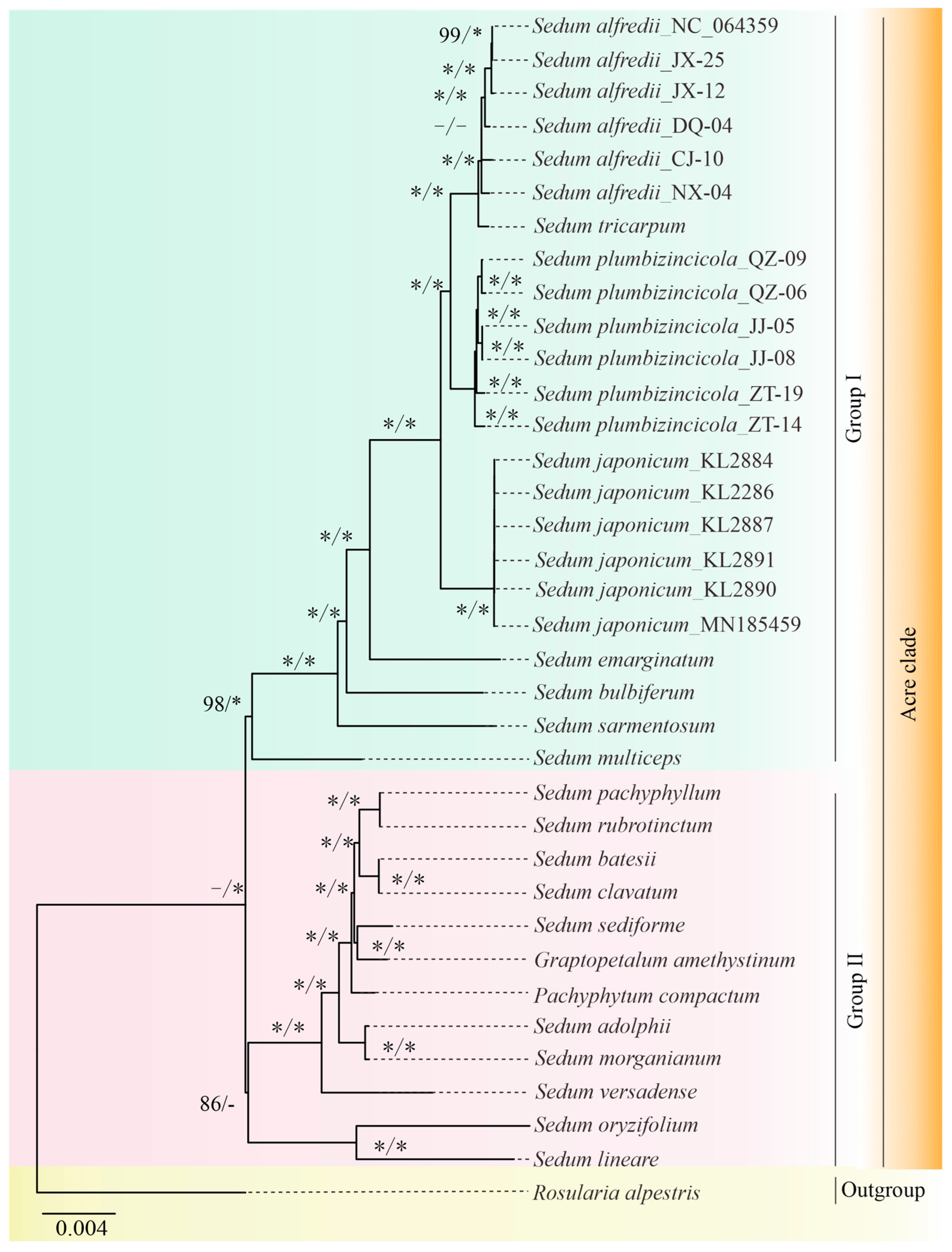
3.4. Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baskin, J.M.; Baskin, C.C. Germination ecology of Sedum pulchellum Michx.(Crassulaceae). Am. J. Bot. 1977, 64, 1242–1247. [Google Scholar] [CrossRef]
- Debabrata, D.; Gupta, J.; Anindya, B. Medicinal exploitation of the plants belonging to the family: Crassulaceae. Nat. Prod. I 2007, 15, 127–135. [Google Scholar]
- Durhman, A.K. Evaluation of Crassulacean Species for Extensive Green Roof Applications; Michigan State University: East Lansing, MI, USA, 2005. [Google Scholar]
- Villarreal, E.L.; Bengtsson, L. Response of a Sedum green-roof to individual rain events. Ecol. Eng. 2005, 25, 1–7. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Z.; Su, Y.; Wang, T. Comparative genomic analysis of Polypodiaceae chloroplasts reveals fine structural features and dynamic insertion sequences. BMC Plant Biol. 2021, 21, 31. [Google Scholar] [CrossRef]
- Peron, F.; De Maria, M.; Spinazzè, F.; Mazzali, U. An analysis of the urban heat island of Venice mainland. Sustain. Cities Soc. 2015, 19, 300–309. [Google Scholar] [CrossRef]
- Gong, Y.; Yin, D.; Fang, X.; Zhai, D.; Li, J. Rainwater retention effect of extensive green roofs monitored under natural rainfall events–a case study in Beijing. Hydrol. Res. 2018, 49, 1773–1787. [Google Scholar] [CrossRef]
- Azkorra, Z.; Pérez, G.; Coma, J.; Cabeza, L.F.; Burés, S.; Álvaro, J.E.; Erkoreka, A.; Urrestarazu, M. Evaluation of green walls as a passive acoustic insulation system for buildings. Appl. Acoust. 2015, 89, 46–56. [Google Scholar] [CrossRef]
- Bi, D.; Han, S.; Zhou, J.; Zhao, M.; Zhang, S.; Kan, X. Codon Usage Analyses Reveal the Evolutionary Patterns among Plastid Genes of Saxifragales at a Larger-Sampling Scale. Genes 2023, 14, 694. [Google Scholar] [CrossRef]
- Ding, H.; Han, S.; Ye, Y.; Bi, D.; Zhang, S.; Yi, R.; Gao, J.; Yang, J.; Wu, L.; Kan, X. Ten plastomes of Crassula (Crassulaceae) and phylogenetic implications. Biology 2022, 11, 1779. [Google Scholar] [CrossRef]
- Messerschmid, T.F.E.; Klein, J.T.; Kadereit, G.; Kadereit, J.W. Linnaeus’s folly—Phylogeny, evolution and classification of Sedum (Crassulaceae) and Crassulaceae subfamily Sempervivoideae. Taxon 2020, 69, 892–926. [Google Scholar] [CrossRef]
- Carrillo-Reyes, P.; Sosa, V.; Mort, M.E. Molecular phylogeny of the Acre clade (Crassulaceae): Dealing with the lack of definitions for Echeveria and Sedum. Mol. Phylogenetics Evol. 2009, 53, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Huang, K.; Xie, P.; Xie, S.; Zhang, N.; Yin, H.; Ping, M.; Wang, Y. The complete chloroplast genome sequence of Sedum bulbiferum (Crassulaceae). Mitochondrial DNA Part B 2023, 8, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. 2015, 90, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ye, Y.; Yi, R.; Bi, D.; Zhang, S.; Han, S.; Kan, X. A new perspective on codon usage, selective pressure, and phylogenetic implications of the plastomes in the Telephium clade (Crassulaceae). Gene 2024, 892, 147871. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwongse, J.; Obokata, J.; Yamaguchi-Shinozaki, K.J.T.E.J.; et al. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 1986, 5, 2043–2049. [Google Scholar] [CrossRef]
- Han, S.; Bi, D.; Yi, R.; Ding, H.; Wu, L.; Kan, X. Plastome evolution of Aeonium and Monanthes (Crassulaceae): Insights into the variation of plastomic tRNAs, and the patterns of codon usage and aversion. Planta 2022, 256, 35. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Guo, J.; Zhou, X.; Ma, K.; Ma, Y.; Shi, T.; Shi, Y. Comparative and evolutionary analyses on the complete plastomes of five Kalanchoe horticultural plants. Front. Plant Sci. 2021, 12, 705874. [Google Scholar] [CrossRef]
- Yu, X.; Wei, P.; Chen, Z.; Li, X.; Zhang, W.; Yang, Y.; Liu, C.; Zhao, S.; Li, X.; Liu, X. Comparative analysis of the organelle genomes of three Rhodiola species provide insights into their structural dynamics and sequence divergences. BMC Plant Biol. 2023, 23, 156. [Google Scholar] [CrossRef]
- Zhao, D.-N.; Ren, C.-Q.; Zhang, J.-Q. Can plastome data resolve recent radiations Rhodiola (Crassulaceae) as a case study. Bot. J. Linn. Soc. 2021, 197, 513–526. [Google Scholar] [CrossRef]
- Zhao, D.-N.; Ren, Y.; Zhang, J.-Q. Conservation and innovation: Plastome evolution during rapid radiation of Rhodiola on the Qinghai-Tibetan Plateau. Mol. Phylogenetics Evol. 2020, 144, 106713. [Google Scholar] [CrossRef]
- Chang, H.; Zhang, L.; Xie, H.; Liu, J.; Xi, Z.; Xu, X. The conservation of chloroplast genome structure and improved resolution of infrafamilial relationships of Crassulaceae. Front. Plant Sci. 2021, 12, 631884. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Zhang, D.Q.; Zhang, J.Q. Plastomic data shed new light on the phylogeny, biogeography, and character evolution of the family Crassulaceae. J. Syst. Evol. 2023, 61, 990–1003. [Google Scholar] [CrossRef]
- Han, S.; Ding, H.; Bi, D.; Zhang, S.; Yi, R.; Gao, J.; Yang, J.; Ye, Y.; Wu, L.; Kan, X. Structural Diversities and Phylogenetic Signals in Plastomes of the Early-Divergent Angiosperms: A Case Study in Saxifragales. Plants 2022, 11, 3544. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. Selection on Codon Bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef]
- Yang, J.; Ding, H.; Kan, X. Codon usage patterns and evolution of HSP60 in birds. Int. J. Biol. Macromol. 2021, 183, 1002–1012. [Google Scholar] [CrossRef]
- Miller, J.B.; McKinnon, L.M.; Whiting, M.F.; Ridge, P.G. CAM: An alignment-free method to recover phylogenies using codon aversion motifs. PeerJ 2019, 7, e6984. [Google Scholar] [CrossRef] [PubMed]
- Aboul-Maaty, N.A.-F.; Oraby, H.A.-S. Extraction of high-quality genomic DNA from different plant orders applying a modified CTAB-based method. Bull. Natl. Res. Cent. 2019, 43, 25. [Google Scholar] [CrossRef]
- McCoy, R.C.; Taylor, R.W.; Blauwkamp, T.A.; Kelley, J.L.; Kertesz, M.; Pushkarev, D.; Petrov, D.A.; Fiston-Lavier, A.S. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PLoS ONE 2014, 9, e106689. [Google Scholar] [CrossRef]
- Nair, S.S.; Luu, P.-L.; Qu, W.; Maddugoda, M.; Huschtscha, L.; Reddel, R.; Chenevix-Trench, G.; Toso, M.; Kench, J.G.; Horvath, L.G. Guidelines for whole genome bisulphite sequencing of intact and FFPET DNA on the Illumina HiSeq X Ten. Epigenetics Chromatin 2018, 11, 24. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; DePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Poczai, P.; Hyvönen, J.; Tang, J.; Amiryousefi, A. Chloroplot: An online program for the versatile plotting of organelle genomes. Front. Genet. 2020, 11, 576124. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 2000. [Google Scholar]
- Sharp, P.M.; Li, W.-H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Cauz-Santos, L.A.; da Costa, Z.P.; Callot, C.; Cauet, S.; Zucchi, M.I.; Berges, H.; van den Berg, C.; Vieira, M.L.C. A Repertory of Rearrangements and the Loss of an Inverted Repeat Region in Passiflora Chloroplast Genomes. Genome Biol. Evol. 2020, 12, 1841–1857. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Bhatt, B.S.; Awasthi, M.; George, B.; Singh, A.K. Comparative analysis of microsatellites in chloroplast genomes of lower and higher plants. Curr. Genet. 2015, 61, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Wang, L.; Lei, J.; Duan, B.; Ma, W.; Xiao, S.; Qi, H.; Wang, Z.; Liu, Y.; Shen, X. A comparative analysis of the chloroplast genomes of four Salvia medicinal plants. Engineering 2019, 5, 907–915. [Google Scholar] [CrossRef]
- Zhu, M.; Feng, P.; Ping, J.; Li, J.; Su, Y.; Wang, T. Phylogenetic significance of the characteristics of simple sequence repeats at the genus level based on the complete chloroplast genome sequences of Cyatheaceae. Ecol. Evol. 2021, 11, 14327–14340. [Google Scholar] [CrossRef]
- Gao, R.; Wang, W.; Huang, Q.; Fan, R.; Wang, X.; Feng, P.; Zhao, G.; Bian, S.; Ren, H.; Chang, Y. Complete chloroplast genome sequence of Dryopteris fragrans (L.) Schott and the repeat structures against the thermal environment. Sci. Rep. 2018, 8, 16635. [Google Scholar] [CrossRef]
- Alawfi, M.S.; Albokhari, E.J. Comparative chloroplast genomics reveals a unique gene inversion in two Cordia trees (Cordiaceae). Forests 2023, 14, 1778. [Google Scholar] [CrossRef]
- Huang, X.; Tan, W.; Li, F.; Liao, R.; Guo, Z.; Shi, T.; Gao, Z. The chloroplast genome of Prunus zhengheensis: Genome comparative and phylogenetic relationships analysis. Gene 2021, 793, 145751. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Wang, L.; Li, Y.; Zhuo, W.; Xu, Z.; Guo, H.; Liu, Y.; Gao, R.; Song, J. Highly variable chloroplast genome from two endangered Papaveraceae lithophytes Corydalis tomentella and Corydalis saxicola. Ecol. Evol. 2021, 11, 4158–4171. [Google Scholar] [CrossRef] [PubMed]
- Vieira, L.d.N.; Faoro, H.; Rogalski, M.; Fraga, H.P.d.F.; Cardoso, R.L.A.; de Souza, E.M.; de Oliveira Pedrosa, F.; Nodari, R.O.; Guerra, M.P. The complete chloroplast genome sequence of Podocarpus lambertii: Genome structure, evolutionary aspects, gene content and SSR detection. PLoS ONE 2014, 9, e90618. [Google Scholar] [CrossRef]
- Yermagambetova, M.; Abugalieva, S.; Turuspekov, Y.; Almerekova, S. Illumina sequencing data of the complete chloroplast genome of rare species Juniperus seravschanica (Cupressaceae) from Kazakhstan. Data Brief 2023, 46, 108866. [Google Scholar] [CrossRef]
- Ebert, D.; Peakall, R. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 2009, 9, 673–690. [Google Scholar] [CrossRef]
- Chen, L.; Liu, X.; Wang, Z.; Wu, X.; Hong, K.; Xie, C. Comparative Chloroplast Genome Analyses of Six Hemlock Trees in East Asia: Insights into Their Genomic Characterization and Phylogenetic Relationship. Forests 2023, 14, 2136. [Google Scholar] [CrossRef]
- Shi, W.; Song, W.; Liu, J.; Shi, C.; Wang, S. Comparative chloroplast genome analysis of Citrus (Rutaceae) species: Insights into genomic characterization, phylogenetic relationships, and discrimination of subgenera. Sci. Hortic. 2023, 313, 111909. [Google Scholar] [CrossRef]
- Wang, Z.; Cai, Q.; Wang, Y.; Li, M.; Wang, C.; Wang, Z.; Jiao, C.; Xu, C.; Wang, H.; Zhang, Z. Comparative analysis of codon Bias in the chloroplast genomes of Theaceae species. Front. Genet. 2022, 13, 824610. [Google Scholar] [CrossRef]
- Dai, Y.; Cui, K.; Duan, M.; Nie, Y.; Jiang, H.; Deng, Y.; He, A.; Chen, J. Differences of Cadmium Extraction Characteristics between Sedum alfredii H. and Sedum plumbizincicola. Res. Sq. 2023. preprint. [Google Scholar]
- Huangrong, Z.; Lianghua, Z.; Feng, Y.; Shiqi, L.; Qiuxiang, Z. Phytoremediation effects and contrast of Sedum alfredii and Sedum plumbizincicola on Cd/Zn contaminated soil. Ecol. Environ. Sci. 2019, 28, 403–410. [Google Scholar]
- Song, W.; Wang, J.; Zhai, L.; Ge, L.; Hao, S.; Shi, L.; Lian, C.; Chen, C.; Shen, Z.; Chen, Y. A meta-analysis about the accumulation of heavy metals uptake by Sedum alfredii and Sedum plumbizincicola in contaminated soil. Int. J. Phytoremediation 2022, 24, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.e.; Long, X.; Ni, W.; Fu, C. Sedum alfredii H: A new Zn hyperaccumulating plant first found in China. Chin. Sci. Bull. 2002, 47, 1634–1637. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Source/Voucher | Accession Number | Locality | GC Content (%) | Length (bp) |
|---|---|---|---|---|---|
| S. alfredii | CJ-10 | PP234500 * | Chunjian, Hangzhou, China | 37.77 | 149,279 |
| (149,401 ± 63 bp) | DQ-04 | PP234501 * | Dongqiao, Hangzhou, China | 37.75 | 149,453 |
| JX-12 | PP234502 * | Jiuxi, Hangzhou, China | 37.76 | 149,427 | |
| JX-25 | PP234503 * | Jiuxi, Hangzhou, China | 37.76 | 149,428 | |
| NX-04 | PP234504 * | Ningxi, Taizhou, China | 37.77 | 149,392 | |
| Shaocui et al. (2022) | NC_064359 | Daqiao, Hangzhou, China | 37.76 | 149,427 | |
| S. japonicum | KL2884 | PP234494 * | Dashan, Chizhou, China | 37.71 | 149,373 |
| (149,377 ± 10 bp) | KL2886 | PP234495 * | Dashan, Chizhou, China | 37.71 | 149,373 |
| KL2887 | PP234496 * | Dashan, Chizhou, China | 37.71 | 149,373 | |
| KL2890 | PP234497 * | Dashan, Chizhou, China | 37.71 | 149,373 | |
| KL2891 | PP234498 * | Dashan, Chizhou, China | 37.71 | 149,373 | |
| Ding et al. (2019) | MN185459 | Dashan, Chizhou, China | 37.71 | 149,397 | |
| S. plumbizincicola | JJ-05 | PP234493 * | Jiangjia, Hangzhou, China | 37.77 | 149,328 |
| (149,316 ± 112 bp) | JJ-08 | PP234488 * | Jiangjia, Hangzhou, China | 37.77 | 149,288 |
| QZ-06 | PP234489 * | Shangfang, Quzhou, China | 37.78 | 149,392 | |
| QZ-09 | PP234490 * | Shangfang, Quzhou, China | 37.77 | 149,394 | |
| ZT-14 | PP234491 * | Zitong, Hangzhou, China | 37.79 | 149,105 | |
| ZT-19 | PP234492 * | Zitong, Hangzhou, China | 37.78 | 149,387 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Han, S.; Bi, D.; Yang, J.; Ge, W.; Ye, Y.; Gao, J.; Dai, C.; Kan, X. Intraspecific and Intrageneric Genomic Variation across Three Sedum Species (Crassulaceae): A Plastomic Perspective. Genes 2024, 15, 444. https://doi.org/10.3390/genes15040444
Zhang S, Han S, Bi D, Yang J, Ge W, Ye Y, Gao J, Dai C, Kan X. Intraspecific and Intrageneric Genomic Variation across Three Sedum Species (Crassulaceae): A Plastomic Perspective. Genes. 2024; 15(4):444. https://doi.org/10.3390/genes15040444
Chicago/Turabian StyleZhang, Sijia, Shiyun Han, De Bi, Jianke Yang, Wen Ge, Yuanxin Ye, Jinming Gao, Chenwei Dai, and Xianzhao Kan. 2024. "Intraspecific and Intrageneric Genomic Variation across Three Sedum Species (Crassulaceae): A Plastomic Perspective" Genes 15, no. 4: 444. https://doi.org/10.3390/genes15040444
APA StyleZhang, S., Han, S., Bi, D., Yang, J., Ge, W., Ye, Y., Gao, J., Dai, C., & Kan, X. (2024). Intraspecific and Intrageneric Genomic Variation across Three Sedum Species (Crassulaceae): A Plastomic Perspective. Genes, 15(4), 444. https://doi.org/10.3390/genes15040444