
Fingerprint Finder: Identifying Genomic Fingerprint Sites in Cotton Cohorts for Genetic Analysis and Breeding Advancement


 and
and 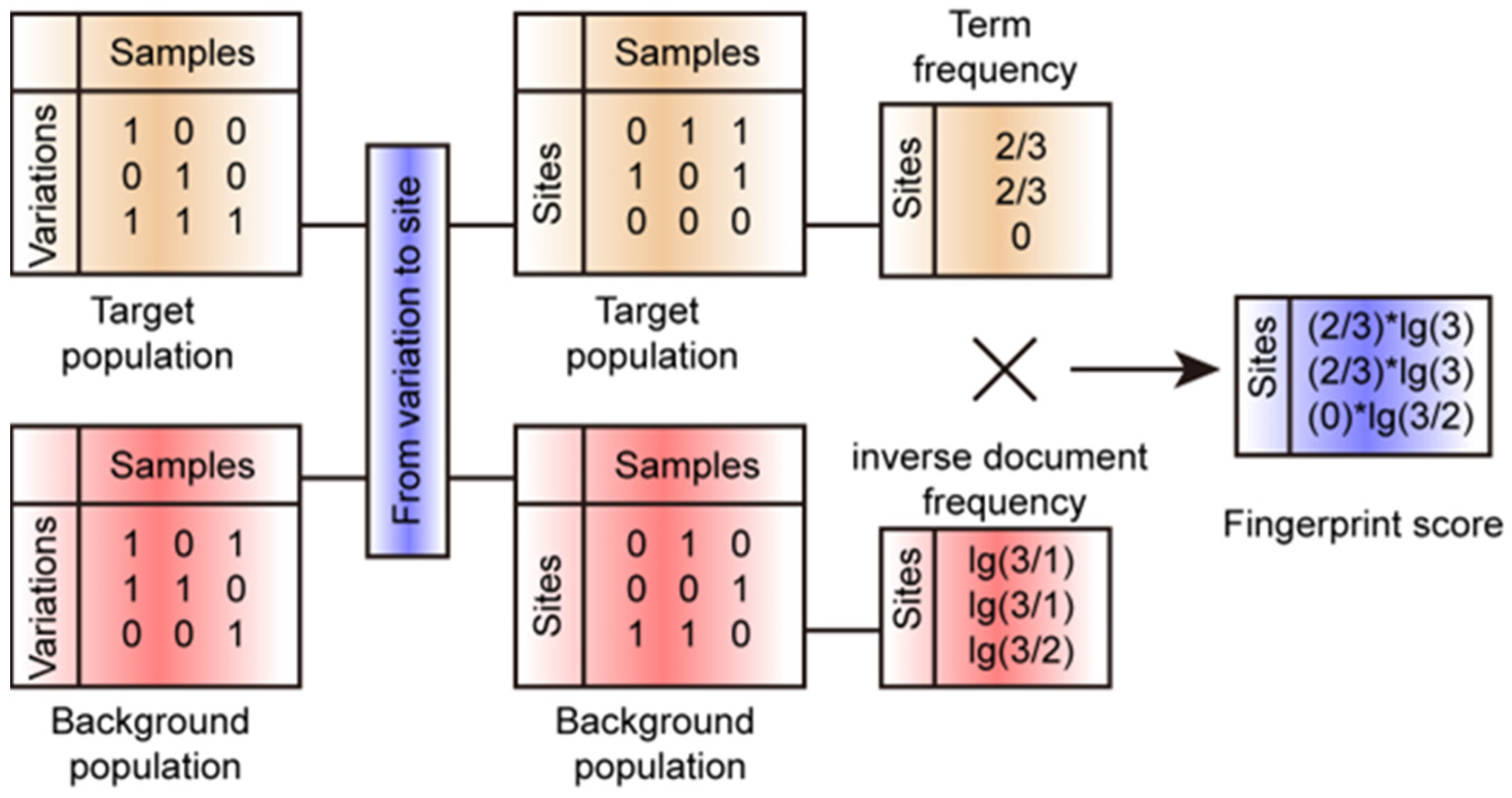{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction, Library Construction and Sequencing
2.3. Data Filtering, Reads Mapping and Variant Calling
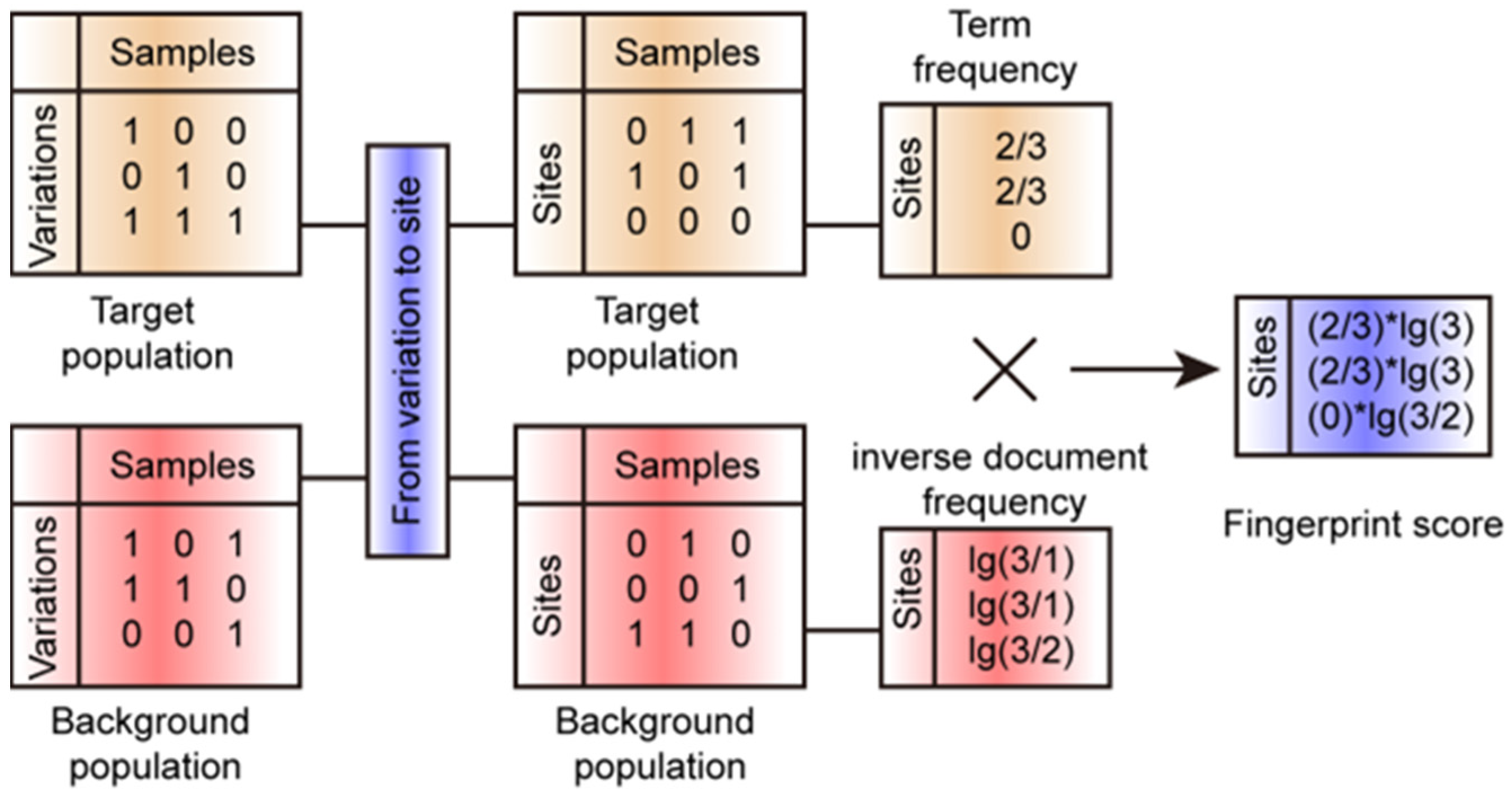
2.4. FPFinder Development
2.5. GWAS Analysis
2.6. Transcriptome Analysis
2.7. WGCNA Pipeline
2.8. Regulatory Network Construction
2.9. Construction of Phenotype Predicting Model
2.10. KEGG Enrichment
2.11. Statistical Analysis and Data Visualization
2.12. qRT-PCR Experiment
3. Results
3.1. Developing the FPFinder
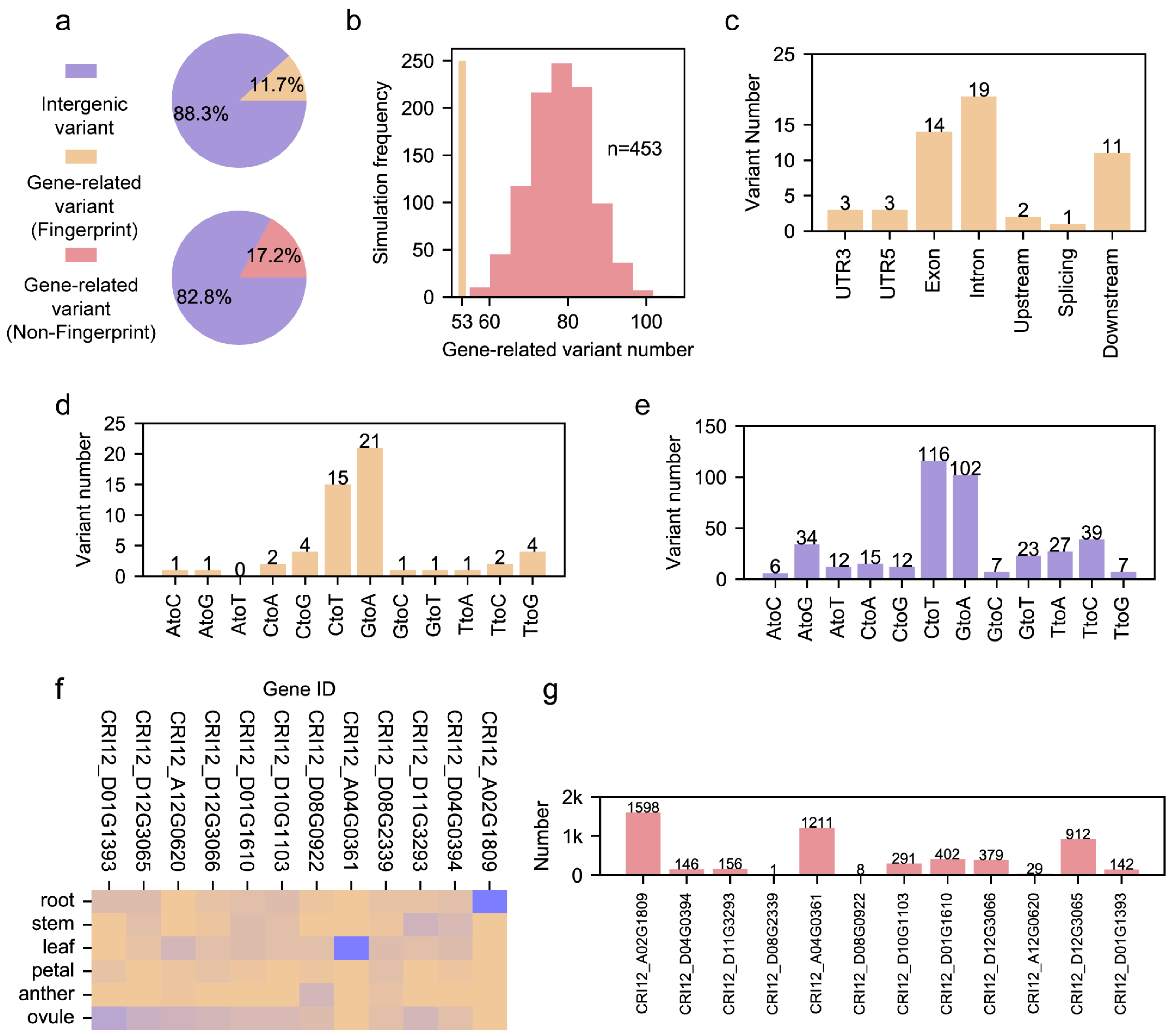
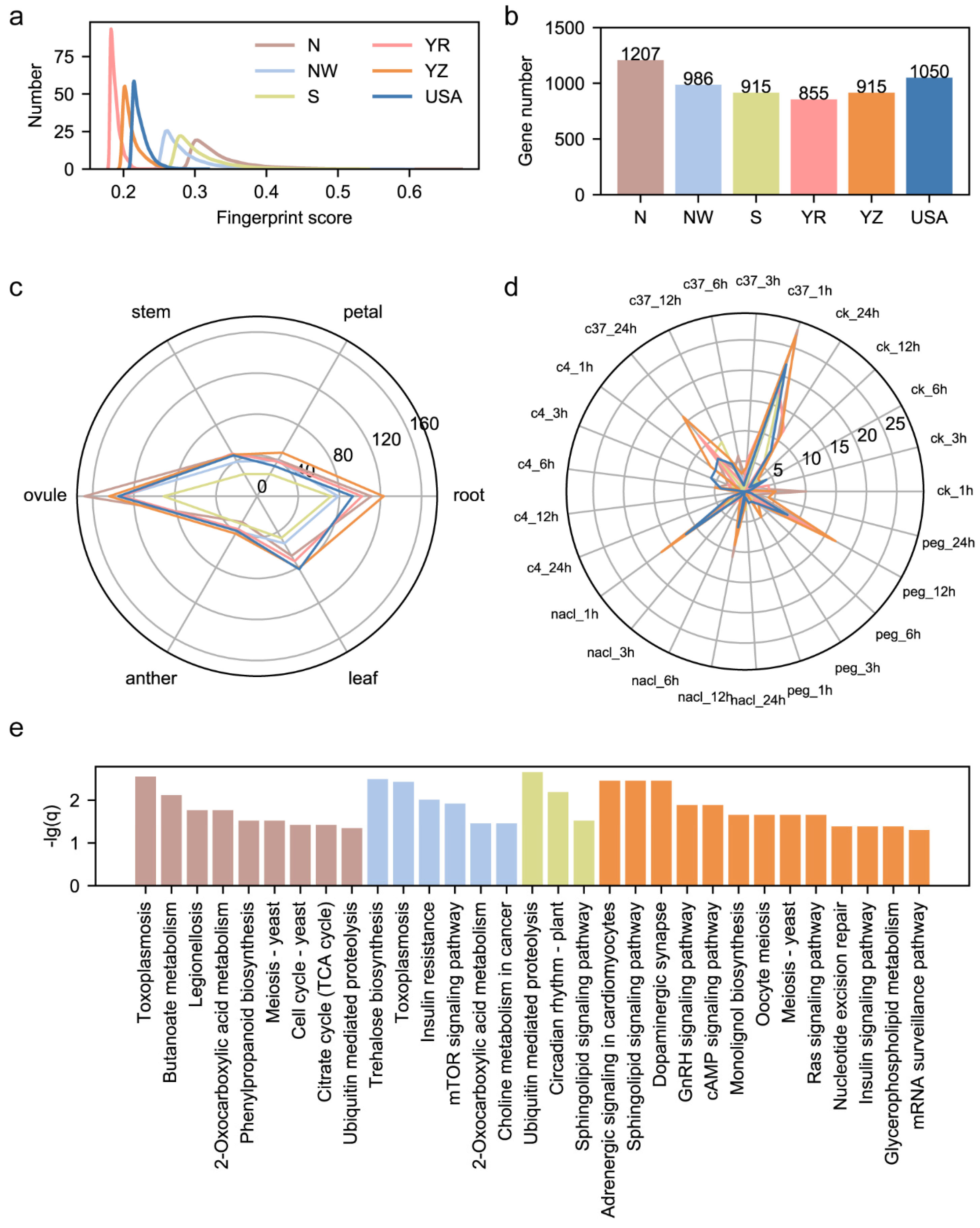
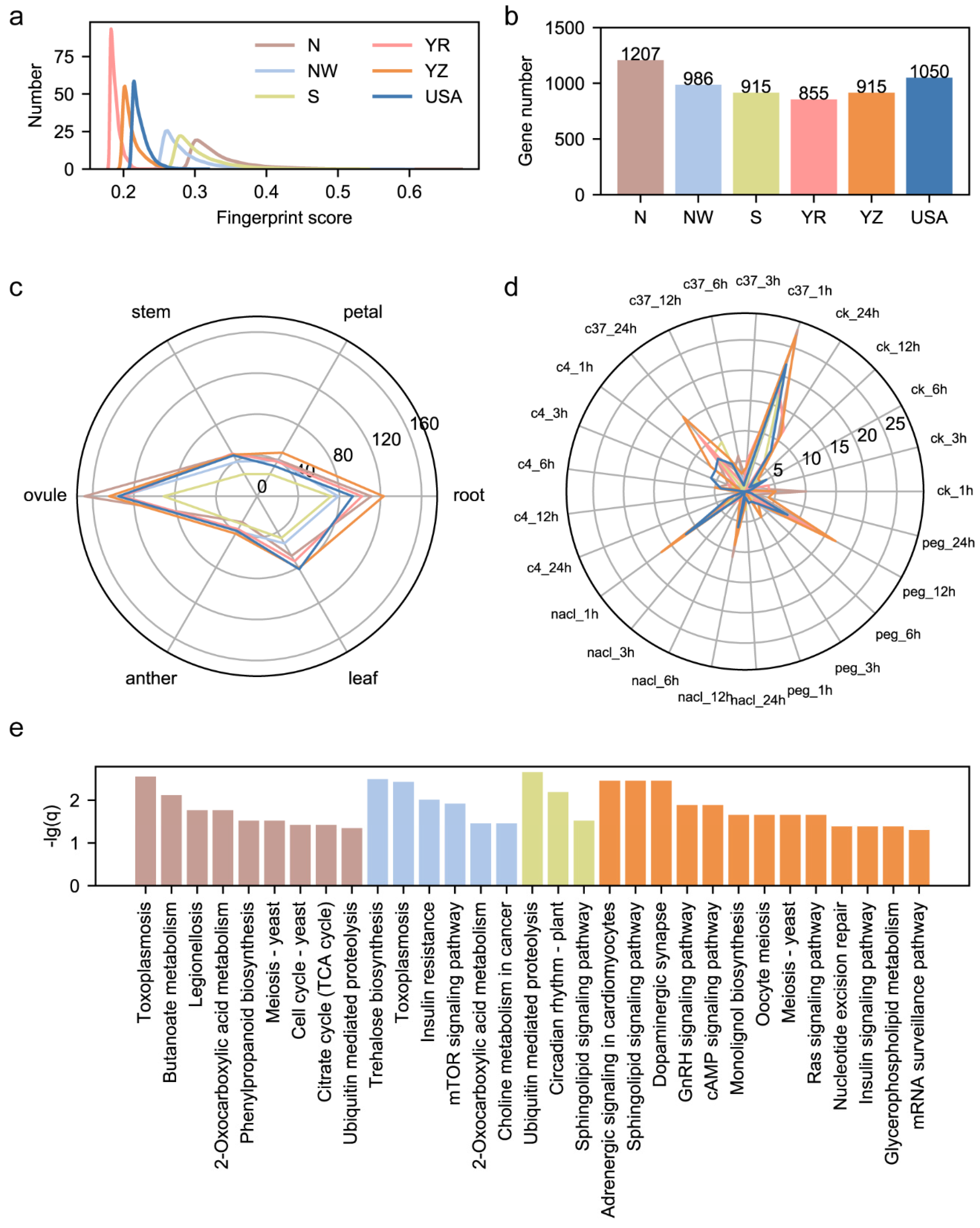
3.2. Characterization of Fingerprint Genomic Sites in Cotton Pedigree
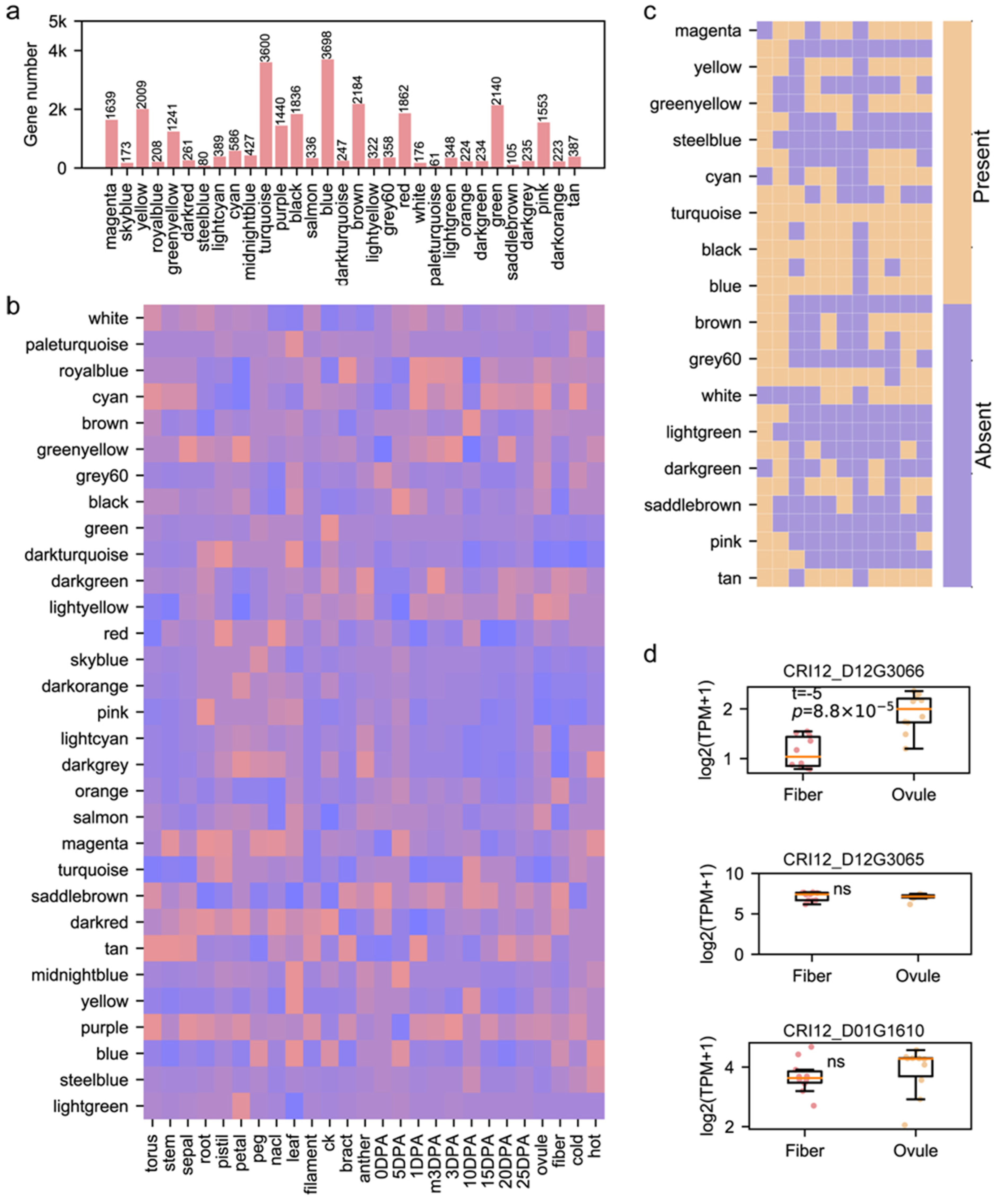
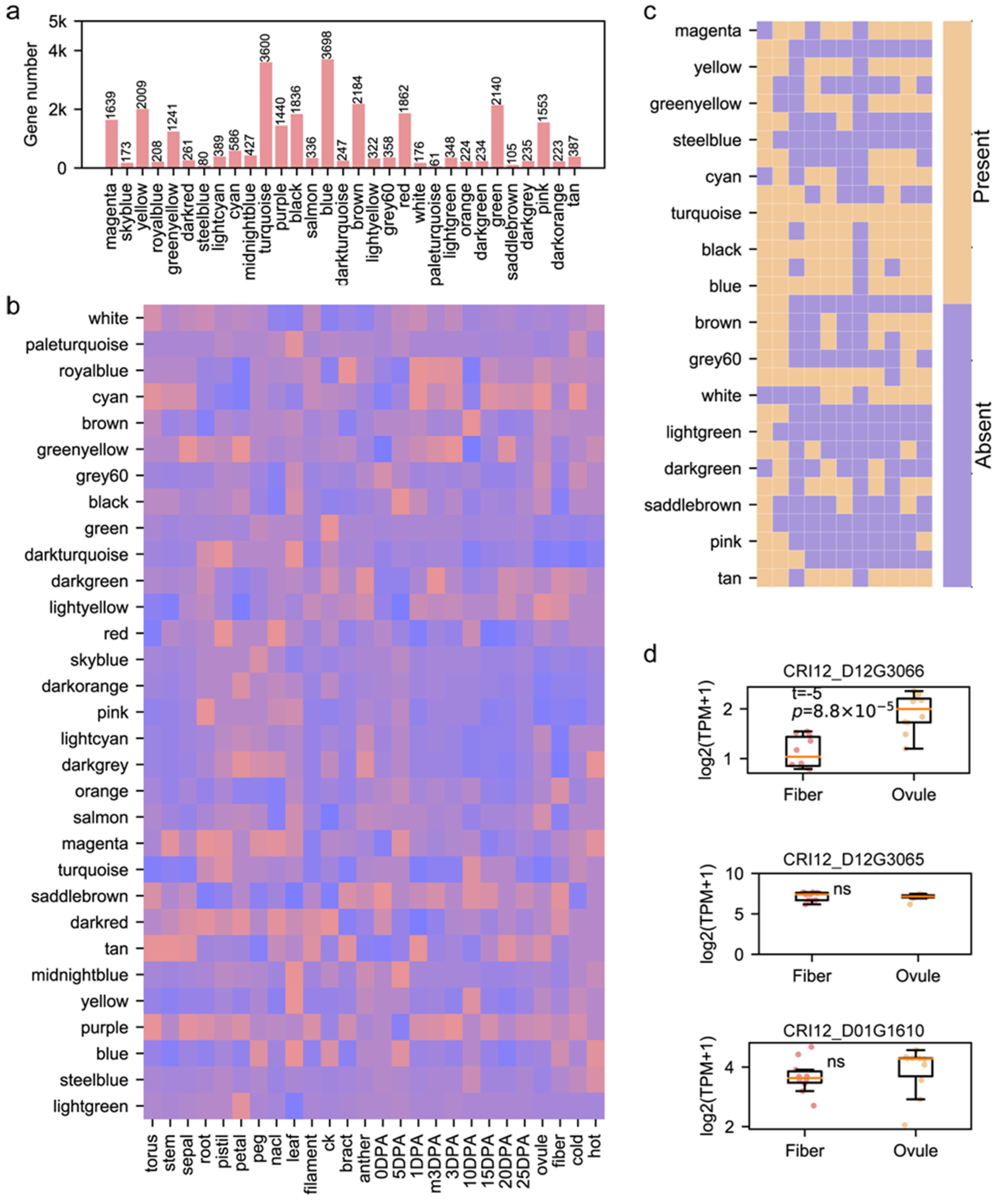
3.3. Role of Pedigree Fingerprint Site-Related Genes in Cotton Development
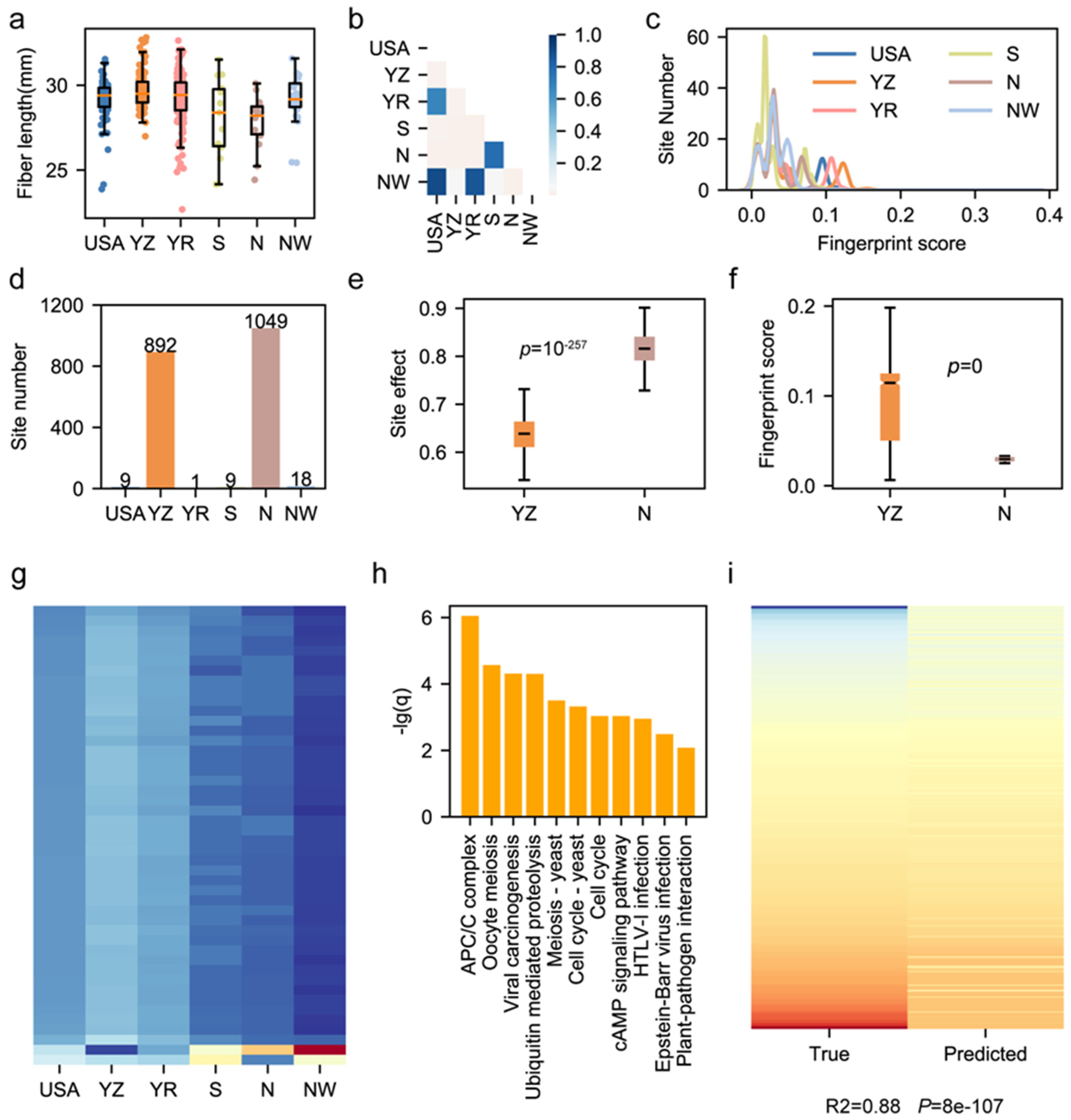
3.4. Geographical Bias of Functional Sites Results in Fiber Length Divergence
3.5. Role of Region-Specific Genomic Sites in Cotton Development
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, Z.; Qanmber, G.; Wang, Z.; Yang, Z.; Li, F. Gossypium Genomics: Trends, Scope, and Utilization for Cotton Improvement. Trends Plant Sci. 2020, 25, 488–500. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Liu, Z.; Qi, Z.; Ma, Y.; Sun, M.; Su, L.; Niu, H.; Peng, Y.; Luo, X.; Zhu, M.; et al. Regulatory controls of duplicated gene expression during fiber development in allotetraploid cotton. Nat. Genet. 2023, 55, 1987–1997. [Google Scholar] [CrossRef]
- Wang, M.; Li, J.; Qi, Z.; Long, Y.; Pei, L.; Huang, X.; Grover, C.E.; Du, X.; Xia, C.; Wang, P.; et al. Genomic innovation and regulatory rewiring during evolution of the cotton genus Gossypium. Nat. Genet. 2022, 54, 1959–1971. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhang, Y.; Wu, L.; Zhang, G.; Sun, Z.; Li, Z.; Jiang, Y.; Ke, H.; Chen, B.; Liu, Z.; et al. High-quality genome assembly and resequencing of modern cotton cultivars provide resources for crop improvement. Nat. Genet. 2021, 53, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, D.; Wang, P.; Wang, Q.; Sun, M.; Liu, Z.; Si, H.; Xu, Z.; Ma, Y.; Zhang, B.; et al. Cotton pan-genome retrieves the lost sequences and genes during domestication and selection. Genome Biol. 2021, 22, 119. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sun, G.; Geng, X.; Gong, W.; Dai, P.; Jia, Y.; Shi, W.; Pan, Z.; Wang, J.; Wang, L.; et al. The genomic basis of geographic differentiation and fiber improvement in cultivated cotton. Nat. Genet. 2021, 53, 916–924. [Google Scholar] [CrossRef]
- Liu, S.; Zuo, D.; Cheng, H.; He, M.; Wang, Q.; Lv, L.; Zhang, Y.; Ashraf, J.; Liu, J.; Song, G. Cotton pedigree genome reveals restriction of cultivar-driven strategy in cotton breeding. Genome Biol. 2023, 24, 282. [Google Scholar] [CrossRef]
- Li, Y.; Si, Z.; Wang, G.; Shi, Z.; Chen, J.; Qi, G.; Jin, S.; Han, Z.; Gao, W.; Tian, Y.; et al. Genomic insights into the genetic basis of cotton breeding in China. Mol. Plant 2023, 16, 662–677. [Google Scholar] [CrossRef]
- Ma, X.; Wang, Z.; Li, W.; Zhang, Y.; Zhou, X.; Liu, Y.; Ren, Z.; Pei, X.; Zhou, K.; Zhang, W.; et al. Resequencing core accessions of a pedigree identifies derivation of genomic segments and key agronomic trait loci during cotton improvement. Plant Biotechnol. J. 2019, 17, 762–775. [Google Scholar] [CrossRef]
- Lu, X.; Fu, X.; Wang, D.; Wang, J.; Chen, X.; Hao, M.; Wang, J.; Gervers, K.A.; Guo, L.; Wang, S.; et al. Resequencing of cv CRI-12 family reveals haplotype block inheritance and recombination of agronomically important genes in artificial selection. Plant Biotechnol. J. 2019, 17, 945–955. [Google Scholar] [CrossRef]
- Sun, X.; Xiang, Y.; Dou, N.; Zhang, H.; Pei, S.; Franco, A.V.; Menon, M.; Monier, B.; Ferebee, T.; Liu, T.; et al. The role of transposon inverted repeats in balancing drought tolerance and yield-related traits in maize. Nat. Biotechnol. 2023, 41, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; Depristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, P.; You, C.; Yu, J.; Zhang, X.; Yan, F.; Ye, Z.; Shen, C.; Li, B.; Guo, K.; et al. Combined GWAS and eQTL analysis uncovers a genetic regulatory network orchestrating the initiation of secondary cell wall development in cotton. New Phytol 2020, 226, 1738–1752. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Liang, C.; Meng, Z.; Sun, G.; Meng, Z.; Guo, S.; Zhang, R. CottonFGD: An integrated functional genomics database for cotton. BMC Plant Biol. 2017, 17, 101. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, D.; Hu, Y.; Li, H.; Ramstein, G.P.; Zhou, S.; Zhang, X.; Bao, Z.; Zhang, Y.; Song, B.; et al. Phylogenomic discovery of deleterious mutations facilitates hybrid potato breeding. Cell 2023, 186, 2313–2328. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Z.; Bao, Z.; Li, H.; Lyu, Y.; Zan, Y.; Wu, Y.; Cheng, L.; Fang, Y.; Wu, K.; et al. Graph pangenome captures missing heritability and empowers tomato breeding. Nature 2022, 606, 527–534. [Google Scholar] [CrossRef]
- Qin, P.; Lu, H.; Du, H.; Wang, H.; Chen, W.; Chen, Z.; He, Q.; Ou, S.; Zhang, H.; Li, X.; et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 2021, 184, 3542–3558. [Google Scholar] [CrossRef]
- Song, J.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-Genome of Wild and Cultivated Soybeans. Cell 2020, 182, 162–176. [Google Scholar] [CrossRef]
- Renna, L.; Stefano, G.; Slabaugh, E.; Wormsbaecher, C.; Sulpizio, A.; Zienkiewicz, K.; Brandizzi, F. TGNap1 is required for microtubule-dependent homeostasis of a subpopulation of the plant trans-Golgi network. Nat. Commun. 2018, 9, 5313. [Google Scholar] [CrossRef]
- Chen, X.; Chern, M.; Canlas, P.E.; Jiang, C.; Ruan, D.; Cao, P.; Ronald, P.C. A conserved threonine residue in the juxtamembrane domain of the XA21 pattern recognition receptor is critical for kinase autophosphorylation and XA21-mediated immunity. J. Biol. Chem. 2010, 285, 10454–10463. [Google Scholar] [CrossRef]
- Qu, X.; Chatty, P.R.; Roeder, A.H. Endomembrane trafficking protein SEC24A regulates cell size patterning in Arabidopsis. Plant Physiol. 2014, 166, 1877–1890. [Google Scholar] [CrossRef]
- Conger, R.; Chen, Y.; Fornaciari, S.; Faso, C.; Held, M.A.; Renna, L.; Brandizzi, F. Evidence for the involvement of the Arabidopsis SEC24A in male transmission. J. Exp. Bot. 2011, 62, 4917–4926. [Google Scholar] [CrossRef]
- Li, S.; Esterberg, R.; Lachance, V.; Ren, D.; Radde-Gallwitz, K.; Chi, F.; Parent, J.L.; Fritz, A.; Chen, P. Rack1 is required for Vangl2 membrane localization and planar cell polarity signaling while attenuating canonical Wnt activity. Proc. Natl. Acad. Sci. USA 2011, 108, 2264–2269. [Google Scholar] [CrossRef]
- Mizutani, M. Impacts of diversification of cytochrome P450 on plant metabolism. Biol. Pharm. Bull. 2012, 35, 824–832. [Google Scholar] [CrossRef]
- Hansen, C.C.; Nelson, D.R.; Møller, B.L.; Werck-Reichhart, D. Plant cytochrome P450 plasticity and evolution. Mol. Plant 2021, 14, 1244–1265. [Google Scholar] [CrossRef] [PubMed]
- Minerdi, D.; Savoi, S.; Sabbatini, P. Role of Cytochrome P450 Enzyme in Plant Microorganisms’ Communication: A Focus on Grapevine. Int. J. Mol. Sci. 2023, 24, 4695. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Guo, Y.; Sun, Q.; Zeng, W.; Li, J.; Li, X.; Xu, W. Genome-Wide Identification of R2R3-MYB Transcription Factors Regulating Secondary Cell Wall Thickening in Cotton Fiber Development. Plant Cell Physiol. 2019, 60, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; He, X.; Tu, L.; Zhu, L.; Zhu, S.; Ge, Z.; Zhang, X. GhJAZ2 negatively regulates cotton fiber initiation by interacting with the R2R3-MYB transcription factor GhMYB25-like. Plant J. 2016, 88, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Li, Q.; Fan, X.; Yang, W.; Xue, Y. The R2R3 MYB transcription factor GhMYB109 is required for cotton fiber development. Genetics 2008, 180, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhu, Y.; Zhang, T. The R3-MYB gene GhCPC negatively regulates cotton fiber elongation. PLoS ONE 2015, 10, e116272. [Google Scholar] [CrossRef]
- Jin, S.; Han, Z.; Hu, Y.; Si, Z.; Dai, F.; He, L.; Cheng, Y.; Li, Y.; Zhao, T.; Fang, L.; et al. Structural variation (SV)-based pan-genome and GWAS reveal the impacts of SVs on the speciation and diversification of allotetraploid cottons. Mol. Plant 2023, 16, 678–693. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Li, Y.; Shen, C.; Yang, Y.; Wang, H.; Yao, T.; Zhang, X.; Lindsey, K.; Lin, Z. High-resolution sequencing of nine elite upland cotton cultivars uncovers genic variations and breeding improvement targets. Plant J. 2023, 113, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Brower-Toland, B.; Shyu, C.; Vega-Sanchez, M.E.; Slewinski, T.L. Pedigree or identity? How genome editing can fundamentally change the path for crop development. J. Exp. Bot. 2023, 74, 2794–2798. [Google Scholar] [CrossRef] [PubMed]
- Van Tassel, D.L.; Dehaan, L.R.; Diaz-Garcia, L.; Hershberger, J.; Rubin, M.J.; Schlautman, B.; Turner, K.; Miller, A.J. Re-imagining crop domestication in the era of high throughput phenomics. Curr. Opin. Plant Biol. 2022, 65, 102150. [Google Scholar] [CrossRef] [PubMed]
- Pikunova, A.; Goryunova, S.; Goryunov, D.; Golyaeva, O.; Dolzhikova, M.; Pavlenko, A. Genetic Diversity and Pedigree Analysis of Red Currant Germplasm. Plants 2022, 11, 1623. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Anumalla, M.; Catolos, M.; Bhosale, S.; Jarquin, D.; Hussain, W. Optimizing predictions in IRRI’s rice drought breeding program by leveraging 17 years of historical data and pedigree information. Front. Plant Sci. 2022, 13, 983818. [Google Scholar] [CrossRef] [PubMed]
- Fradgley, N.; Gardner, K.A.; Cockram, J.; Elderfield, J.; Hickey, J.M.; Howell, P.; Jackson, R.; Mackay, I.J. A large-scale pedigree resource of wheat reveals evidence for adaptation and selection by breeders. PLoS Biol. 2019, 17, e3000071. [Google Scholar] [CrossRef] [PubMed]
- Braun, B.L.; Schott, D.A.; Portwood, J.L.; Andorf, C.M.; Sen, T.Z. PedigreeNet: A web-based pedigree viewer for biological databases. Bioinformatics 2019, 35, 4184–4186. [Google Scholar] [CrossRef]
- Paik, H.; Cho, Y.; Cho, S.B.; Kwon, O.K. MPI-GWAS: A supercomputing-aided permutation approach for genomewide association studies. Genom. Inform. 2022, 20, e14. [Google Scholar] [CrossRef]
- John, M.; Ankenbrand, M.J.; Artmann, C.; Freudenthal, J.A.; Korte, A.; Grimm, D.G. Efficient permutation-based genome-wide association studies for normal and skewed phenotypic distributions. Bioinformatics 2022, 38, i5–i12. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wen, J.; Eckert, S.T.; Wang, Y.; Liu, D.J.; Wu, R.; Li, R.; Zhan, X. Prioritizing genetic variants in GWAS with lasso using permutation-assisted tuning. Bioinformatics 2020, 36, 3811–3817. [Google Scholar] [CrossRef] [PubMed]
- Leem, S.; Huh, I.; Park, T. Enhanced Permutation Tests via Multiple Pruning. Front. Genet. 2020, 11, 509. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Wen, X.; Chen, Z.; Yang, Z.; Wang, M.; Jin, S.; Wang, G.; Zhang, L.; Wang, L.; Li, J.; Saeed, S.; et al. A comprehensive overview of cotton genomics, biotechnology and molecular biological studies. Sci. China Life Sci. 2023, 66, 2214–2256. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Cheng, H.; Zhang, Y.; He, M.; Zuo, D.; Wang, Q.; Lv, L.; Lin, Z.; Song, G. Fingerprint Finder: Identifying Genomic Fingerprint Sites in Cotton Cohorts for Genetic Analysis and Breeding Advancement. Genes 2024, 15, 378. https://doi.org/10.3390/genes15030378
Liu S, Cheng H, Zhang Y, He M, Zuo D, Wang Q, Lv L, Lin Z, Song G. Fingerprint Finder: Identifying Genomic Fingerprint Sites in Cotton Cohorts for Genetic Analysis and Breeding Advancement. Genes. 2024; 15(3):378. https://doi.org/10.3390/genes15030378
Chicago/Turabian StyleLiu, Shang, Hailiang Cheng, Youping Zhang, Man He, Dongyun Zuo, Qiaolian Wang, Limin Lv, Zhongxv Lin, and Guoli Song. 2024. "Fingerprint Finder: Identifying Genomic Fingerprint Sites in Cotton Cohorts for Genetic Analysis and Breeding Advancement" Genes 15, no. 3: 378. https://doi.org/10.3390/genes15030378
APA StyleLiu, S., Cheng, H., Zhang, Y., He, M., Zuo, D., Wang, Q., Lv, L., Lin, Z., & Song, G. (2024). Fingerprint Finder: Identifying Genomic Fingerprint Sites in Cotton Cohorts for Genetic Analysis and Breeding Advancement. Genes, 15(3), 378. https://doi.org/10.3390/genes15030378