Genomic Characterization of SARS-CoV-2 Variants from Clinical Isolates during the COVID-19 Epidemic in Mauritania

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Site
2.2. Sample Collection
2.3. Viral RNA Extraction and Ampslification
2.4. SARS-CoV-2 Whole Genome Sequencing
2.5. Data Analysis
3. Results
3.1. Characteristics of Clinical Isolates
3.2. Patient Characteristics
3.3. SARS-CoV-2 Genome Sequence Analysis and Variants
3.4. Characteristic Mutations in the Spike Protein of Delta and Eta Variants
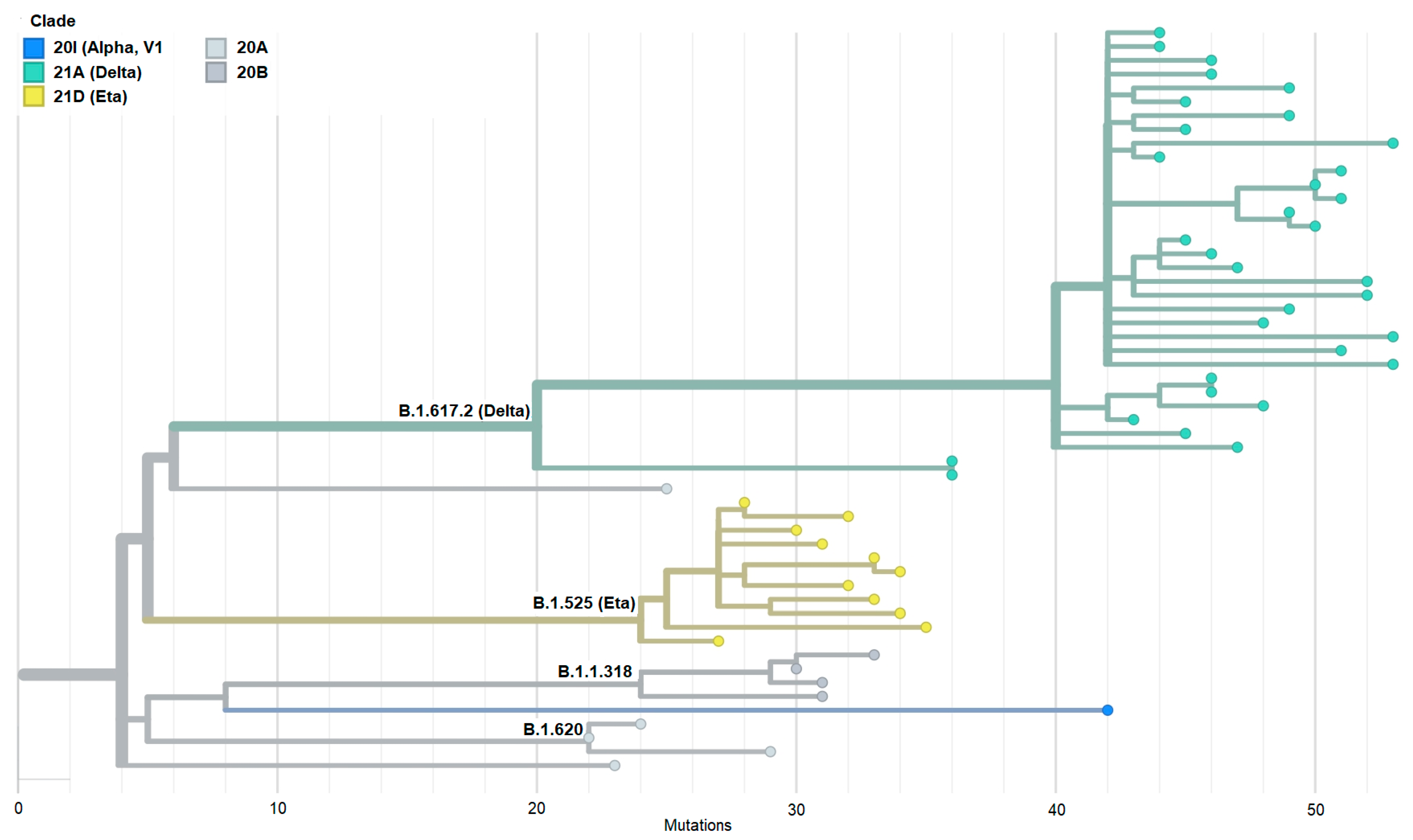
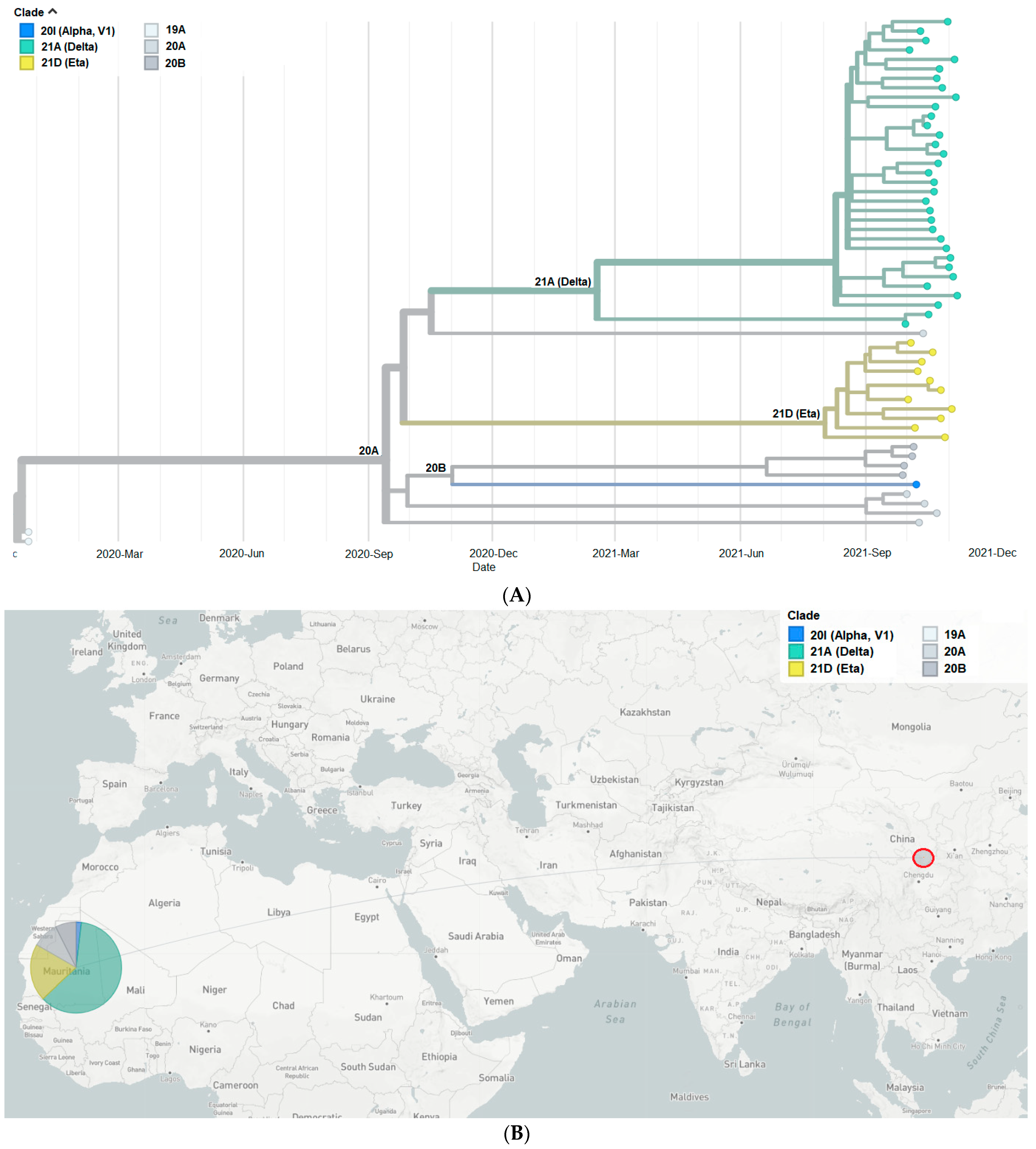
3.5. Phylogeny of SARS-CoV-2 Isolates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zhu, H.; Wei, L.; Niu, P. The novel coronavirus outbreak in Wuhan, China. Glob. Health Res. Policy 2020, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- WHO. Coronavirus Disease (COVID-19) Pandemic. Available online: https://www.who.int/europe/emergencies/situations/covid-19 (accessed on 3 March 2024).
- WHO. COVID-19 Epidemiological Update: 22 December 2023 (Edition 162). Available online: https://www.who.int/publications/m/item/covid-19-epidemiological-update---22-december-2023 (accessed on 25 January 2024).
- WHO. Over Two-Thirds of Africans Exposed to Virus Which Causes COVID-19: WHO Study. WHO Regional Office for Africa, 2022. Available online: https://www.afro.who.int/news/over-two-thirds-africans-exposed-virus-which-causes-covid-19-who-study (accessed on 3 March 2024).
- Lalaoui, R.; Bakour, S.; Raoult, D.; Verger, P.; Sokhna, C.; Devaux, C.; Pradines, B.; Rolain, J.M. What could explain the late emergence of COVID-19 in Africa? New Microbes New Infect. 2020, 38, 100760. [Google Scholar] [CrossRef]
- China National Center for Bioinformation. RCoV19—Variation Annotation. 2024. Available online: https://ngdc.cncb.ac.cn/ncov/variation/annotation (accessed on 11 February 2024).
- Sgorlon, G.; Roca, T.P.; Passos-Silva, A.M.; Custódio, M.G.F.; Queiroz, J.A.D.S.; da Silva, A.L.F.; Teixeira, K.S.; Batista, F.S.; Salcedo, J.M.V.; Rampazzo, R.C.P.; et al. SARS-CoV-2 spike protein mutations in different variants: A comparison between vaccinated and unvaccinated population in western Amazonia. Bioinform. Biol. Insights 2023, 17, 11779322231186477. [Google Scholar] [CrossRef]
- Guruprasad, K. Mutations in human SARS-CoV-2 spike proteins, potential drug binding and epitope sites for COVID-19 therapeutics development. Curr. Res. Struct. Biol. 2022, 4, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- DeGrace, M.M.; Ghedin, E.; Frieman, M.B.; Krammer, F.; Grifoni, A.; Alisoltani, A.; Alter, G.; Amara, R.R.; Baric, R.S.; Barouch, D.H.; et al. Defining the risk of SARS-CoV-2 variants on immune protection. Nature 2022, 605, 640–652. [Google Scholar] [CrossRef]
- Zhao, S.; Lou, J.; Cao, L.; Chong, K.C.; Zee, B.C.Y.; Chan, P.K.S.; Wang, M.H. Differences in the case fatality risks associated with SARS-CoV-2 Delta and non-Delta variants in relation to vaccine coverage: An early ecological study in the United Kingdom. Infect. Genet. Evol. 2022, 97, 105162. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.; Poduri, R. Omicron, a new SARS-CoV-2 variant: Assessing the impact on severity and vaccines efficacy. Hum. Vaccin. Immunother. 2022, 18, 2034458. [Google Scholar] [CrossRef]
- Wang, C.; Liu, B.; Zhang, S.; Huang, N.; Zhao, T.; Lu, Q.B.; Cui, F. Differences in incidence and fatality of COVID-19 by SARS-CoV-2 Omicron variant versus Delta variant in relation to vaccine coverage: A world-wide review. J. Med. Virol. 2023, 95, e28118. [Google Scholar] [CrossRef]
- El Vally, A.; Bollahi, M.A.; Ould Ahmedou Salem, M.S.; Deida, J.; Parola, P.; Basco, L.; El Bara, A.; Ouldabdallahi, M.; Ould Mohamed Salem Boukhary, A. Retrospective overview of a COVID-19 outbreak in Mauritania. New Microbes New Infect. 2020, 38, 100788. [Google Scholar] [CrossRef]
- Ahmed, M.L.C.B.; Zehaf, S.; El Alem, M.M.; Elbara, A.; Ely Mahmoud, M.M.; Mohamed Abdellahi, M.V.; Heukelbach, J. COVID-19 outbreak in Mauritania: Epidemiology and health system response. J. Infect. Dev. Ctries. 2021, 15, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Laghdaf, S.M.; El Baraa, A.; Mohamed Beydjeu, A. Characterization of a cluster of COVID-19 cases linked to the Omicron variant, in Mauritania. Tunis. Med. 2022, 100, 217–221. [Google Scholar]
- Abdelmalick, A.; Sehli, S.; Idrissi Azami, A.; Habib, N.; Al Idrissi, N.; Belyamani, L.; Houmeida, A.; Ghazal, H. Genomic evidence of multiple introductions of SARS-CoV-2 in Mauritania. Bioinform. Biol. Insights. 2023, 17, 11779322231167927. [Google Scholar] [CrossRef] [PubMed]
- WHO. Mauritania: The Current COVID-19 Situation. Available online: https://www.who.int/countries/mrt/ (accessed on 25 January 2024).
- Accoe, K.; Criel, B.; Ag Ahmed, M.A.; Buitrago, V.T.; Marchal, B. Conditions for health system resilience in the response to the COVID-19 pandemic in Mauritania. BMJ Glob. Health 2023, 8, e013943. [Google Scholar] [CrossRef]
- Agence Nationale de la Statistique et de l’Analyse Démographique et Economique. 2022. Available online: https://ansade.mr/fr/ (accessed on 3 March 2024).
- Papa Mze, N.; Kacel, I.; Beye, M.; Tola, R.; Sarr, M.; Basco, L.; Bogreau, H.; Colson, P.; Fournier, P.E. High throughput SARS-CoV-2 genome sequencing from 384 respiratory samples using the Illumina COVIDSeq protocol. Genes 2023, 14, 681. [Google Scholar] [CrossRef] [PubMed]
- ARTIC Network. Real-Time Molecular Epidemiology for Outbreak Response. Available online: https://artic.network (accessed on 11 February 2024).
- Garrison, E.; Marth, G. Freebayes, a Haplotype-Based Variant Detector: User Manual and Guide. MIT License, 2010. Available online: https://github.com/freebayes/freebayes (accessed on 29 January 2024).
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907v2. [Google Scholar] [CrossRef]
- Li, H. BWA-MEM2 (Sequence Alignment Using Burrows-Wheeler Transform), v2.2.1. MIT License, 2019. Available online: https://github.com/bwa-mem2/bwa-mem2 (accessed on 12 September 2022).
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. In Proceedings of the 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; pp. 314–324. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Genome Research Ltd. Samtools. Available online: https://github.com/samtools/samtools (accessed on 12 September 2022).
- Li, H.; Handsaker, B.; Danecek, P.; Samtools Team. BCFtools(1) Manual Page. Available online: https://samtools.github.io/bcftools/bcftools.html (accessed on 12 September 2022).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Bedford, T.; Neher, R. Nextclade Web 2.12.0, Nextclade CLI 2.12.0. Copyright 2020–2024. Available online: https://clades.nextstrain.org (accessed on 12 September 2022).
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Centre for Genomic Pathogen Surveillance. Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN). Available online: https://cov-lineages.org/pangolin.html (accessed on 12 September 2022).
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- MedCalc Software Ltd. Fisher Exact Probability Calculator, Version 22.021. Available online: https://www.medcalc.org/calc/fisher.php (accessed on 4 March 2024).
- WHO. Updated Working Definitions and Primary Actions for SARS-CoV-2 Variants, 4 October 2023; World Health Organization: Geneva, Switzerland, 2023; Available online: https://www.who.int/publications/m/item/updated-working-definitions-and-primary-actions-for--sars-cov-2-variants (accessed on 29 January 2024).
- Tregoning, J.S.; Flight, K.E.; Higham, S.L.; Wang, Z.; Pierce, B.F. Progress of the COVID-19 vaccine effort: Viruses, vaccines and variants versus efficacy, effectiveness and escape. Nat. Rev. Immunol. 2021, 21, 626–636. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 spike mutations, L452R, T478K, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef] [PubMed]
- Annavajhala, M.K.; Mohri, H.; Wang, P.; Nair, M.; Zucker, J.E.; Sheng, Z.; Gomez-Simmonds, A.; Kelley, A.L.; Tagliavia, M.; Huang, Y.; et al. Emergence and expansion of SARS-CoV-2 B.1.526 after identification in New York. Nature 2021, 597, 703–708. [Google Scholar] [CrossRef] [PubMed]
- United Kingdom Health Security Agency (UKHSA). SARS-CoV-2 Variant Surveillance and Assessment: Technical Briefing 55. UK Government, 2023. Available online: https://www.gov.uk/gouvernment/publications/investigations-of-sars-cov-2-variants-technical-briefings/sars-cov-2-variant-surveillance-and-assessment-technical-briefing-55 (accessed on 18 February 2024).
- Carpenter, R.E.; Tamrakar, V.K.; Almas, S.; Sharma, A.; Sharma, R. SARS-CoV-2 Next Generation Sequencing (NGS) data from clinical isolates from the East Texas Region of the United States. Data Brief. 2023, 49, 109312. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, A.J.S.; Beye, M.; Sow, A.; Lo, G.; Padane, A.; Sokhna, C.; Kane, C.T.; Colson, P.; Fenollar, F.; Mboup, S.; et al. COVID-19 in 16 West African countries: An assessment of the epidemiology and genetic diversity of SARS-CoV-2 after four epidemic waves. Am. J. Trop. Med. Hyg. 2023, 109, 861–873. [Google Scholar] [CrossRef]
- Tian, D.; Sun, Y.; Zhou, J.; Ye, Q. The global epidemic of the SARS-CoV-2 Delta variant, key spike mutations and immune escape. Front. Immunol. 2021, 12, 751778. [Google Scholar] [CrossRef]
- Li, M.; Lou, F.; Fan, H. SARS-CoV-2 Variants of Concern Delta: A great challenge to prevention and control of COVID-19. Signal Transduct. Target. Ther. 2021, 6, 349. [Google Scholar] [CrossRef]
- Shiehzadegan, S.; Alaghemand, N.; Fox, M.; Venketaraman, V. Analysis of the Delta variant B.1.617.2 COVID-19. Clin. Pract. 2021, 11, 778–784. [Google Scholar] [CrossRef]
- Koné, A.; Diallo, D.; Kané, F.; Diarra, B.; Coulibaly, T.A.; Sameroff, S.C.; Diarra, H.B.; Diakité, M.T.; Camara, F.; Maiga, O.; et al. Dynamics of SARS-CoV-2 variants characterized during different COVID-19 waves in Mali. IJID Reg. 2023, 6, 24–28. [Google Scholar] [CrossRef]
- Padane, A.; Mbow, M.; Mboup, A.; Diedhiou, C.K.; Gueye, K.; Lo, C.I.; Ndiour, S.; Leye, N.; Ndoye, A.S.; Selbé Ndiaye, A.J.; et al. Rapidly rising cases with Omicron in Senegal. New Microbes New Infect. 2022, 45, 100959. [Google Scholar] [CrossRef]
- Yadav, P.D.; Nyayanit, D.A.; Gupta, N.; Shastri, J.; Sahay, R.R.; Patil, D.Y.; Shete, A.M.; Razdan, A.; Agrawal, S.; Kumar, A.; et al. Detection and isolation of SARS-CoV-2 Eta variant from the international travelers and local residents of India. J. Med. Virol. 2022, 94, 3404–3409. [Google Scholar] [CrossRef]
- Park, A.K.; Kim, I.H.; Man Kim, H.; Lee, H.; Lee, N.J.; Kim, J.A.; Woo, S.; Lee, C.Y.; Lee, J.; Oh, S.J.; et al. SARS-CoV-2 B.1.619 and B.1.620 lineages, South Korea, 2021. Emerg. Infect. Dis. 2022, 28, 415–419. [Google Scholar] [CrossRef]
- Manouana, G.P.; Nzamba Maloum, M.; Bikangui, R.; Oye Bingono, S.O.; Ondo Nguema, G.; Honkpehedji, J.Y.; Rossatanga, E.G.; Zoa-Assoumou, S.; Pallerla, S.R.; Rachakonda, S.; et al. Emergence of B.1.1.318 SARS-CoV-2 viral lineage and high incidence of alpha B.1.1.7 variant of concern in the Republic of Gabon. Int. J. Infect. Dis. 2022, 114, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Singanayagam, A.; Hakki, S.; Dunning, J.; Madon, K.J.; Crone, M.A.; Koycheva, A.; Derqui-Fernandez, N.; Barnett, J.L.; Whitfield, M.G.; Varro, R.; et al. Community transmission and viral load kinetics of the SARS-CoV-2 delta (B.1.617.2) variant in vaccinated and unvaccinated individuals in the UK: A prospective, longitudinal, cohort study. Lancet Infect. Dis. 2022, 22, 183–195, Erratum in Lancet Infect. Dis. 2021, 21, e363. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.R.; Jiang, Y.W.; Li, F.X.; Liu, D.; Lin, T.F.; Zhao, Z.Y.; Wei, C.; Jin, Q.Y.; Li, X.M.; Jia, Y.X.; et al. Efficacy of SARS-CoV-2 vaccines and the dose-response relationship with three major antibodies: A systematic review and meta-analysis of randomised controlled trials. Lancet Microbe. 2023, 4, e236–e246. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Ye, Q. The variants of SARS-CoV-2 and the challenges of vaccines. J. Med. Virol. 2022, 94, 1366–1372. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| WHO Classification * | Clade | Pango Lineage | WHO Label | Origin | Frequency (%) |
|---|---|---|---|---|---|
| VOC | 21J | AY.34 (alias B.1.617.2.34) | Delta | Multiple countries | 31 (57.4) |
| 21I | AY.61 (alias B.1.617.2.61) | Delta | Italy | 1 (1.9) | |
| AY.75 (alias B.1.617.2.75) | Delta | USA and Europe | 1 (1.9) | ||
| 20I | B.1.1.7 | Alpha | United Kingdom | 1 (1.9) | |
| 20B | B.1.1.528 | 20B | South Africa | 1 (1.9) | |
| 20A | B.1 | 20A | Italy | 1 (1.9) | |
| VOI | 21D | B.1.525 | Eta | Nigeria | 11 (20.4) |
| 20B | AZ.3 | 20B | USA | 1 (1.9) | |
| 20B | B.1.1.318 | 20B | Multiple countries | 3 (5.5) | |
| VUM | 20A | B.1.620 | 20A | Potentially from Cameroon | 3 (5.5) |
| Variant or Clade | Vaccinated | Unvaccinated |
|---|---|---|
| VOC | 21 | 15 |
| VOI + VUM | 12 | 6 |
| Total | 33 | 21 |
| Clade AY.34 | 18 | 13 |
| Other clades | 15 | 8 |
| Total | 33 | 21 |
| Variant * | AA ** Substitution | No. Isolates (%) | Reported Effect of the Mutation | Reference |
|---|---|---|---|---|
| Delta (n = 33) | T19R | 32 (97) |
| [37,38] |
| L452R | 30 (91) | |||
| T478K | 32 (97) | |||
| D614G | 33 (100) | |||
| P681R | 33 (100) | |||
| D950N | 33 (100) | |||
| Eta (n = 9) | Q52R | 9 (100) |
| [39,40] |
| E484K | 8 (88.9) | |||
| D614G | 9 (100) | |||
| Q677H | 9(100) | |||
| F888L | 8 (88.9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deida, J.; Papa Mze, N.; Beye, M.; Ahmed, S.M.; El Bara, A.; Bollahi, M.A.; Basco, L.; Ould Mohamed Salem Boukhary, A.; Fournier, P.-E. Genomic Characterization of SARS-CoV-2 Variants from Clinical Isolates during the COVID-19 Epidemic in Mauritania. Genes 2024, 15, 361. https://doi.org/10.3390/genes15030361
Deida J, Papa Mze N, Beye M, Ahmed SM, El Bara A, Bollahi MA, Basco L, Ould Mohamed Salem Boukhary A, Fournier P-E. Genomic Characterization of SARS-CoV-2 Variants from Clinical Isolates during the COVID-19 Epidemic in Mauritania. Genes. 2024; 15(3):361. https://doi.org/10.3390/genes15030361
Chicago/Turabian StyleDeida, Jemila, Nasserdine Papa Mze, Mamadou Beye, Sidi Mohamed Ahmed, Ahmed El Bara, Mohamed Abdallahi Bollahi, Leonardo Basco, Ali Ould Mohamed Salem Boukhary, and Pierre-Edouard Fournier. 2024. "Genomic Characterization of SARS-CoV-2 Variants from Clinical Isolates during the COVID-19 Epidemic in Mauritania" Genes 15, no. 3: 361. https://doi.org/10.3390/genes15030361
APA StyleDeida, J., Papa Mze, N., Beye, M., Ahmed, S. M., El Bara, A., Bollahi, M. A., Basco, L., Ould Mohamed Salem Boukhary, A., & Fournier, P.-E. (2024). Genomic Characterization of SARS-CoV-2 Variants from Clinical Isolates during the COVID-19 Epidemic in Mauritania. Genes, 15(3), 361. https://doi.org/10.3390/genes15030361