
Massive Parallel DNA Sequencing of Patients with Inherited Cardiomyopathies in Cyprus and Suggestion of Digenic or Oligogenic Inheritance

 , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Data and Blood Samples from Patients
2.2. Targeted Massive Parallel DNA Sequencing
- A.
- Probands with cardiomyopathies
- B.
- Individuals of the Cypriot general population
2.3. Bioinformatics Analysis of the Next Generation Sequencing Data
- A.
- Probands with cardiomyopathies
- Functional consequences: missense, nonsense, frameshift, in frame coding indels, and splice site (±2 nucleotides) variants;
- MAF ≤ 0.03 (per the 1000 Genomes Project Consortium);
- Classified as pathogenic, likely pathogenic, and variants of uncertain significance (VUS) in the Franklin tool (based on the ACMG criteria).
- B.
- Individuals of the Cypriot general population
2.4. Sanger DNA Sequencing
3. Results
3.1. Clinical Data of Patients
3.2. Genetic Investigations and DNA Mutation Findings
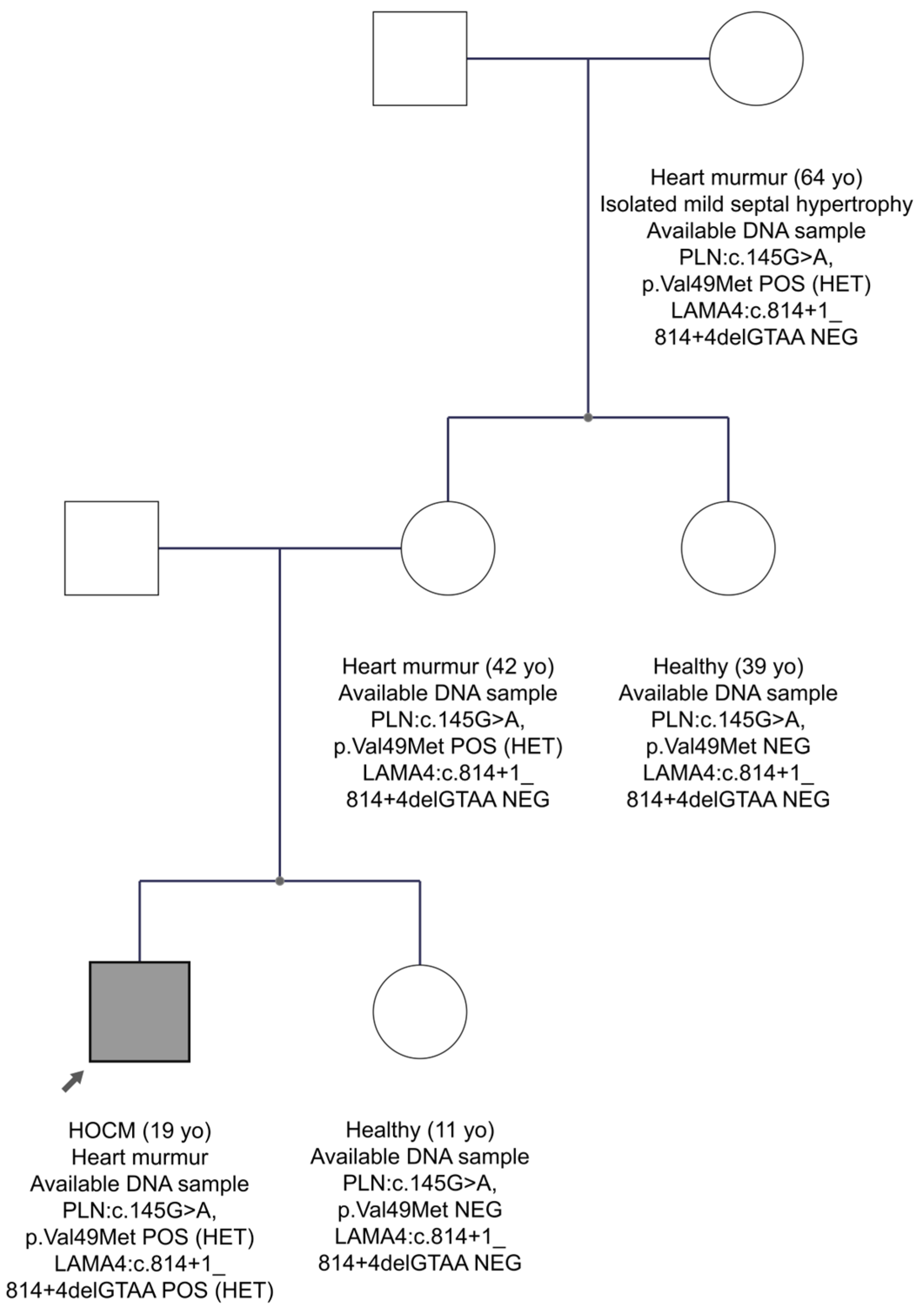
3.3. Family Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; Association American Heart; et al. Contemporary definitions and classification of the cardiomyopathies: An american heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 2006, 113, 1807–1816. [Google Scholar]
- Bezzina, C.R.; Lahrouchi, N.; Priori, S.G. Genetics of sudden cardiac death. Circ. Res. 2015, 116, 1919–1936. [Google Scholar] [CrossRef]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A molecular basis for familial hypertrophic cardiomyopathy: A β cardiac myosin heavy chain gene missense mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- Seidman, C.E.; Seidman, J.G. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy a personal history. Circ. Res. 2011, 108, 743–750. [Google Scholar] [CrossRef]
- Teekakirikul, P.; Zhu, W.; Huang, H.C.; Fung, E. Hypertrophic cardiomyopathy: An overview of genetics and management. Biomolecules 2019, 9, 878. [Google Scholar] [CrossRef]
- Kamisago, M.; Sharma, S.D.; DePalma, S.R.; Solomon, S.; Sharma, P.; McDonough, B.; Smoot, L.; Mullen, M.P.; Woolf, P.K.; Wigle, E.D.; et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 2000, 343, 1688–1696. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef]
- McNally, E.M.; Mestroni, L. Dilated cardiomyopathy: Genetic determinants and mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- Syrris, P.; Ward, D.; Evans, A.; Asimaki, A.; Gandjbakhch, E.; Sen-Chowdhry, S.; McKenna, W.J. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am. J. Hum. Genet. 2006, 79, 978–984. [Google Scholar] [CrossRef]
- Hoorntje, E.T.; Rijdt, W.P.T.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; van Tintelen, J.P. Arrhythmogenic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Herren, T.; Gerber, P.A.; Duru, F. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: A not so rare “disease of the desmosome” with multiple clinical presentations. Clin. Res. Cardiol. 2009, 98, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Miszalski-Jamka, K.; Jefferies, J.L.; Mazur, W.; Glowacki, J.; Hu, J.; Lazar, M.; Gibbs, R.A.; Liczko, J.; Klys, J.; Venner, E.; et al. Novel genetic triggers and genotype-phenotype correlations in patients with left ventricular noncompaction. Circ. Cardiovasc. Genet. 2017, 10, e001763. [Google Scholar] [CrossRef] [PubMed]
- Moric-Janiszewska, E.; Markiewicz-Loskot, G. Genetic heterogeneity of left-ventricular noncompaction cardiomyopathy. Clin. Cardiol. 2008, 31, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, J.; Kubo, T.; Duque, M.; Uribe, W.; Shaw, A.; Murphy, R.; Gimeno, J.R.; Elliott, P.; McKenna, W.J. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin i mutations. J. Clin. Investig. 2003, 111, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Kaski, J.P.; Syrris, P.; Burch, M.; Tome-Esteban, M.T.; Fenton, M.; Christiansen, M.; Andersen, P.S.; Sebire, N.; Ashworth, M.; Deanfield, J.E.; et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 2008, 94, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Kostareva, A.; Kiselev, A.; Gudkova, A.; Frishman, G.; Ruepp, A.; Frishman, D.; Smolina, N.; Tarnovskaya, S.; Nilsson, D.; Zlotina, A.; et al. Genetic spectrum of idiopathic restrictive cardiomyopathy uncovered by next-generation sequencing. PLoS ONE 2016, 11, e0163362. [Google Scholar] [CrossRef] [PubMed]
- Dorn, C.; Grunert, M.; Sperling, S.R. Application of high-throughput sequencing for studying genomic variations in congenital heart disease. Brief. Funct. Genom. 2014, 13, 51–65. [Google Scholar] [CrossRef]
- Ware, S.M.; Jefferies, J.L. New genetic insights into congenital heart disease. J. Clin. Exp. Cardiol. 2012, S8, 3. [Google Scholar] [CrossRef]
- Harris, P.A.; Taylor, R.; Minor, B.L.; Elliott, V.; Fernandez, M.; O’Neal, L.; McLeod, L.; Delacqua, G.; Delacqua, F.; Kirby, J.; et al. The redcap consortium: Building an international community of software platform partners. J. Biomed. Inform. 2019, 95, 103208. [Google Scholar] [CrossRef]
- Girdea, M.; Dumitriu, S.; Fiume, M.; Bowdin, S.; Boycott, K.M.; Chenier, S.; Chitayat, D.; Faghfoury, H.; Meyn, M.S.; Ray, P.N.; et al. Phenotips: Patient phenotyping software for clinical and research use. Hum. Mutat. 2013, 34, 1057–1065. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.; Sirotkin, K. Dbsnp-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999, 9, 677–679. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. Clinvar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The human gene mutation database (hgmd((r))): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of samtools and bcftools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Krasheninina, O.; Hwang, Y.C.; Bai, X.; Zalcman, A.; Maxwell, E.; Reid, J.G.; Salerno, W.J., Jr. Open-source mapping and variant calling for large-scale ngs data from original base-quality scores. bioRxiv 2020. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.F.; Rodeh, O.; Penn, J.; Bai, X.; Reid, J.G.; Krasheninina, O.; Salerno, W.J. Glnexus: Joint variant calling for large cohort sequencing. BioRxiv 2018, 343970. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation plink: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Zhao, H.; Sun, Z.; Wang, J.; Huang, H.; Kocher, J.P.; Wang, L. Crossmap: A versatile tool for coordinate conversion between genome assemblies. Bioinformatics 2014, 30, 1006–1007. [Google Scholar] [CrossRef]
- Pagel, K.A.; Kim, R.; Moad, K.; Busby, B.; Zheng, L.; Tokheim, C.; Ryan, M.; Karchin, R. Integrated informatics analysis of cancer-related variants. JCO Clin. Cancer Inform. 2020, 4, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Task Force members; Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; et al. 2014 esc guidelines on diagnosis and management of hypertrophic cardiomyopathy: The task force for the diagnosis and management of hypertrophic cardiomyopathy of the european society of cardiology (esc). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Miner, J.H.; Patton, B.L.; Lentz, S.I.; Gilbert, D.J.; Snider, W.D.; Jenkins, N.A.; Copeland, N.G.; Sanes, J.R. The laminin alpha chains: Expression, developmental transitions, and chromosomal locations of alpha1–5, identification of heterotrimeric laminins 8–11, and cloning of a novel alpha3 isoform. J. Cell Biol. 1997, 137, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Knoll, R.; Postel, R.; Wang, J.; Kratzner, R.; Hennecke, G.; Vacaru, A.M.; Vakeel, P.; Schubert, C.; Murthy, K.; Rana, B.K.; et al. Laminin-α4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation 2007, 116, 515–525. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef]
- Chiu, C.; Tebo, M.; Ingles, J.; Yeates, L.; Arthur, J.W.; Lind, J.M.; Semsarian, C. Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2007, 43, 337–343. [Google Scholar] [CrossRef]
- Xu, J.; Li, Z.; Ren, X.; Dong, M.; Li, J.; Shi, X.; Zhang, Y.; Xie, W.; Sun, Z.; Liu, X.; et al. Investigation of pathogenic genes in chinese sporadic hypertrophic cardiomyopathy patients by whole exome sequencing. Sci. Rep. 2015, 5, 16609. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Ackerman, M.J. Beyond the cardiac myofilament: Hypertrophic cardiomyopathy- associated mutations in genes that encode calcium-handling proteins. Curr. Mol. Med. 2012, 12, 507–518. [Google Scholar] [CrossRef]
- Heuser, A.; Plovie, E.R.; Ellinor, P.T.; Grossmann, K.S.; Shin, J.T.; Wichter, T.; Basson, C.T.; Lerman, B.B.; Sasse-Klaassen, S.; Thierfelder, L.; et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 1081–1088. [Google Scholar] [CrossRef]
- Zhao, Y.; Cao, H.; Song, Y.; Feng, Y.; Ding, X.; Pang, M.; Zhang, Y.; Zhang, H.; Ding, J.; Xia, X. Identification of novel mutations including a double mutation in patients with inherited cardiomyopathy by a targeted sequencing approach using the ion torrent pgm system. Int. J. Mol. Med. 2016, 37, 1511–1520. [Google Scholar] [CrossRef]
- Deltas, C. Digenic inheritance and genetic modifiers. Clin. Genet. 2018, 93, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Deltas, C.; Savva, I.; Voskarides, K.; Papazachariou, L.; Pierides, A. Carriers of autosomal recessive alport syndrome with thin basement membrane nephropathy presenting as focal segmental glomerulosclerosis in later life. Nephron 2015, 130, 271–280. [Google Scholar] [CrossRef]
- Voskarides, K.; Patsias, C.; Pierides, A.; Deltas, C. Col4a3 founder mutations in greek-cypriot families with thin basement membrane nephropathy and focal segmental glomerulosclerosis dating from around 18th century. Genet. Test. 2008, 12, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Deltas, C.; Gale, D.; Cook, T.; Voskarides, K.; Athanasiou, Y.; Pierides, A. C3 glomerulonephritis/cfhr5 nephropathy is an endemic disease in cyprus: Clinical and molecular findings in 21 families. Adv. Exp. Med. Biol. 2013, 734, 189–196. [Google Scholar]
- Stavrou, C.; Koptides, M.; Tombazos, C.; Psara, E.; Patsias, C.; Zouvani, I.; Kyriacou, K.; Hildebrandt, F.; Christofides, T.; Pierides, A.; et al. Autosomal-dominant medullary cystic kidney disease type 1: Clinical and molecular findings in six large cypriot families. Kidney Int. 2002, 62, 1385–1394. [Google Scholar] [CrossRef]
- Bleyer, A.J.; Kmoch, S.; Antignac, C.; Robins, V.; Kidd, K.; Kelsoe, J.R.; Hladik, G.; Klemmer, P.; Knohl, S.J.; Scheinman, S.J.; et al. Variable clinical presentation of an muc1 mutation causing medullary cystic kidney disease type 1. Clin. J. Am. Soc. Nephrol. 2014, 9, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Andreou, S.; Panayiotou, E.; Michailidou, K.; Pirpa, P.; Hadjisavvas, A.; El Salloukh, A.; Barnes, D.; Antoniou, A.; Agathangelou, P.; Papastavrou, K.; et al. Epidemiology of attrv30m neuropathy in cyprus and the modifier effect of complement c1q on the age of disease onset. Amyloid 2018, 25, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The 72-Gene Panel | ||||||||
|---|---|---|---|---|---|---|---|---|
| ABCC9 | CACNA1C | DSC2 | GLA | KCNQ1 | MYL2 | NKX2.5 | RBM20 | TMPO |
| ACTA1 | CACNB2 | DSG2 | HCN4 | LAMA4 | MYL3 | PDLIM3 | RYR2 | TNNC1 |
| ACTC1 | CASQ2 | DSP | JPH2 | LAMP2 | MYLK2 | PKP2 | SCN5A | TNNI3 |
| ACTN2 | CAV3 | DTNA | JUP | LDB3 | MYOM1 | PLN | SGCD | TNNT2 |
| ALPK3 | CRYAB | EMD | KCNE1 | LMNA | MYOZ2 | PRDM16 | TAZ | TPM1 |
| ANK2 | CSRP3 | EYA4 | KCNE2 | MYBPC3 | MYPN | PRKAG2 | TCAP | TTN |
| ANKRD1 | DES | FKTN | KCNH2 | MYH6 | NEBL | PTPN11 | TGFB3 | TTR |
| BAG3 | DMD | FLNC | KCNJ2 | MYH7 | NEXN | RAF1 | TMEM43 | VCL |
| Family Number | Clinical Phenotype | Number of Family Members with Available DNA Samples | Gender of the Proband | Age at Diagnosis of the Proband | Genetic Variants Found in the Proband | Number of Family Members with the Genetic Variants | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | Patients | Healthy | Total | Clinically Affected Carriers | Clinically Healthy Carriers | |||||
| FAM01 | HCM | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 | M | 60 | ALPK3:c.5548A>G, p.Lys1850Glu (HET) | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 |
| FAM02 | HCM | 9 (5 M/4 F) | 1 (1 M/0 F) | 8 (4 M/4 F) | M | 64 | VCL:c.2415_2421delTGGAAAC, p.Gly806PhefsTer47 (HET) | 4 (2 M/2 F) | 1 (1 M/0 F) | 3 (1 M/2 F) |
| FAM03 | HOCM | 2 (2 M/0 F) | 1 (1 M/0 F) | 1 (1 M/0 F) | M | 63 | TNNI3:c.428C>A, p.Thr143Asn (HET) | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 |
| TTN:c.81809_81811delAAG, p.Glu27270del (HET) | 2 (2 M/0 F) | 1 (1 M/0 F) | 1 (1 M/0 F) | |||||||
| FAM04 | HCM | 10 (5 M/5 F) | 2 (0 M/2 F) | 8 (5 M/3 F) | F | 62 | FLNC:c.2635C>T, p.Arg879Cys (HET) | 4 (1 M/3 F) | 1 (0 M/1 F) | 3 (1 M/2 F) |
| MYPN:c.3959T>C, p.Leu1320Pro (HET) | 2 (0 M/2 F) | 1 (0 M/1 F) | 1 (0 M/1 F) | |||||||
| FAM05 | HCM | 2 (1 M/1 F) | 1 (1 M/0 F) | 1 (0 M/1 F) | M | 15 | MYPBC3:c.3697C>T, p.Gln1233Ter (HET) | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 |
| FAM06 | HCM | 9 (6 M/3 F) | 1 (1 M/0 F) | 8 (5 M/3 F) | M | 10 | MYH7:c.1357C>A, p.Arg453Ser (HET) | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 |
| RBM20:c.3584C>A, p.Ser1195Tyr (HET) | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 | |||||||
| FAM07 | HCM | 2 (2 M/0 F) | 1 (1 M/0 F) | 1 (1 M/0 F) | M | 50 | RBM20:c.2761A>G, p.Ile921Val (HET) | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 |
| FAM08 | HOCM | 3 (1 M/2 F) | 2 (0 M/2 F) | 1 (1 M/0 F) | F | 65 | RBM20:c.*1T>G (in the 3-UTR) (HET) * | 2 (0 M/2 F) | 2 (0 M/2 F) | 0 |
| FAM09 | HOCM | 6 (3 M/3 F) | 1 (0 M/1 F) | 5 (3 M/2 F) | F | 60 | KCNQ1:c.1768G>A, p.Ala590Thr (HET) | 2 (1 M/1 F) | 1 (0 M/1 F) | 1 (1 M/0 F) |
| MYH7:c.4985G>A, p.Arg1662His (HET) | 4 (1 M/3 F) | 1 (0 M/1 F) | 3 (1 M/2 F) | |||||||
| DSC2:c.1891A>G, p.Thr631Ala (HET) | 5 (2 M/3 F) | 1 (0 M/1 F) | 4 (2 M/2 F) | |||||||
| FAM10 | HOCM | 5 (1 M/4 F) | 1 (1 M/0 F) | 4 (0 M/4 F) | M | 13 | PLN:c.145G>A, p.Val49Met (HET) | 3 (1 M/2 F) | 1 (1 M/0 F) | 2 (0 M/2 F) |
| LAMA4:c.814+1_814+4delGTAA (HET) | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 | |||||||
| FAM11 | HCM | 4 (3 M/1 F) | 3 (2 M/1 F) | 1 (1 M/0 F) | F | 35 | MYH7:2156G>A, p.Arg719Gln (HET) | 3 (2 M/1 F) | 3 (2 M/1 F) | 0 |
| RYR2:c.9625C>A, p.Pro3209Thr (HET) | 3 (2 M/1 F) | 3 (2 M/1 F) | 0 | |||||||
| FAM12 | HCM | 2 (1 M/1 F) | 2 (1 M/1 F) | 0 | M | 35 | TTN:c.53273G>C, p.Arg17758Pro (HET) | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 |
| TTN:c.35271G>C, p.Glu11757Asp (HET) | 2 (1 M/1 F) | 2 (1 M/1 F) | 0 | |||||||
| FAM13 | HCM | 5 (3 M/2 F) | 1, 3? (2 M/2 F) | 1 (1 M/0 F) | M | 35 | ALPK3:c.4094C>T, p.Ala1365Val (HET) | 3 (2 M/1 F) | 1, 2? (2 M/1 F) | 0 |
| FAM14 | HCM | 3 (2 M/1 F) | 1 (1 M/0 F) | 2 (1 M/1 F) | M | 19 | TTN:c.22718G>T, p.Arg7573Ile (HET) | 2 (2 M/0 F) | 1 (1 M/0 F) | 1 (1 M/0 F) |
| DSP:c.7154G>A, p.Arg2385His (HET) | 2 (1 M/1 F) | 1 (1 M/0 F) | 1 (0 M/1 F) | |||||||
| FAM15 | DCM | 6 (2 M/4 F) | 1 (0 M/1 F) | 5 (2 M/3 F) | F | 65 | ANK2:c.11458C>T, p.Arg3820Trp (HET) | 3 (0 M/3 F) | 1 (0 M/1 F) | 2 (0 M/2 F) |
| DSP:c.5324G>T, p.Arg1775Ile (HET) | 3 (1 M/2 F) | 1 (0 M/1 F) | 2 (1 M/1 F) | |||||||
| FAM16 | DCM | 6 (1 M/5 F) | 1 (0 M/1 F) | 5 (1 M/4 F) | F | 28 | LAMP2:c.3G>C, p.Met1Ile (HET) | 1 (0 M/1 F) | 1 (0 M/1 F) | 0 |
| FAM17 | DCM | 7 (5 M/2 F) | 3 (2 M/1 F) | 4 (3 M/1 F) | F | 38 | NEBL:c.2513T>C, p.Ile838Thr (HET) | 4 (2 M/2 F) | 2 (1 M/1 F) | 2 (1 M/1 F) |
| MYL2:c.359G>A, p.Arg120Gln (HET) | 3 (2 M/1 F) | 2 (1 M/1 F) | 1 (1 M/0 F) | |||||||
| FAM18 | DCM | 2 (2 M/0 F) | 1 (1 M/0 F) | 1 (1 M/0 F) | M | 53 | LMNA:c.908_909delCT, p.Ser303CysfsTer (HET) | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 |
| FAM19 | DCM | 3 (3 M/0 F) | 1 (1 M/0 F) | 2 (2 M/0 F) | M | 19 | MYH7:c.2290T>C, p.Phe764Leu (HET) | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 |
| MYH7:c.4985G>A, p.Arg1662His (HET) | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 | |||||||
| SCN5A:c.5086C>T, p.Leu1696Phe (HET) | 2 (2 M/0 F) | 1 (1 M/0 F) | 1 (1 M/0 F) | |||||||
| FAM20 | DCM | 3 (1 M/2 F) | 1 (1 M/0 F) | 2 (0 M/2 F) | M | 65 | NEXN:c.1582_1584delGAA, p.Glu528del (HET) | 2 (1 M/1 F) | 1 (1 M/0 F) | 1 (0 M/1 F) |
| TTN:c.51560A>C, p.Asn17187Thr (HET) | 2 (1 M/1 F) | 1 (1 M/0 F) | 1 (0 M/1 F) | |||||||
| TTN:c.88802G>A, p.Arg29601His (HET) | 2 (1 M/1 F) | 1 (1 M/0 F) | 1 (0 M/1 F) | |||||||
| FAM21 | DCM | 8 (5 M/3 F) | 1 (0 M/1 F) | 7 (5 M/2 F) | F | 70 | TTN:c.87043_87044insCA, p.Ile29015ThrfsTer15 (HET) | 5 (4 M/1 F) | 1 (0 M/1 F) | 4 (4 M/0 F) |
| PKP2:c.184C>A, p.Gln62Lys (HET) | 5 (3 M/2 F) | 1 (0 M/1 F) | 4 (3 M/1 F) | |||||||
| FAM22 | ARVC | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 | M | 40 | DES:c.128A>C, p.Lys43Thr (HET) | 1 (1 M/0 F) | 1 (1 M/0 F) | 0 |
| FAM23 | ARVC | 4 (2 M/2 F) | 0 | 4 (2 M/2 F) | F | The proband is healthy but she has a positive family history of ARVC | DSC2:c.133delG, p.Ala45ProfsTer10 (HET) | 2 (1 M/1 F) | 0 | 2 (1 M/1 F) |
| MYH6:c.5072G>C, p.Arg1691Pro (HET) | 3 (1 M/2 F) | 0 | 3 (1 M/2 F) | |||||||
| FAM24 | ARVC | 4 (4 M/0 F) | 3 (3 M/0 F) | 1 (1 M/0 F) | M | 20 | DSC2:c.133delG, p.Ala45ProfsTer10 (HET) | 4 (4 M/0 F) | 3 (3 M/0 F) | 1 (1 M/0 F) |
| DSC2:c.991C>A, p.Gln331Lys (HET) | 3 (3 M/0 F) | 3 (3 M/0 F) | 0 (0 M/0 F) | |||||||
| FAM25 | NCM | 5 (3 M/2 F) | 1 (1 M/0 F) | 4 (2 M/2 F) | M | 19 | TNNC1:c.435C>A, p.Asp145Glu (HET) | 2 (2 M/0 F) | 1 (1 M/0 F) | 1 (1 M/0 F) |
| Family Number | Gene (Exon) | Chromosome Position; Transcript | Coding | Protein | dbSNP Entry | MAF (1000 Genomes) | MAF (GnomAD Exomes) | CY-MAF (Based on 1100 Samples) | HGMD Professional | Franklin | ClinVar | Our Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FAM01 (HCM) | ALPK3 (Exon 14) | chr15:85411511; NM_020778.4 | c.5548A>G | p.Lys1850Glu | rs1273857977 | Not reported | Not reported | Not found | Not reported | VUS to likely benign | Not reported | VUS |
| FAM02 (HCM) | VCL (Exon 16) | chr10:75865090; NM_014000.2 | c.2415_2421delTGGAAAC | p.Gly806PhefsTer47 | Not reported | Not reported | Not reported | 0.000454545 | Not reported | Likely pathogenic | Not reported | Likely pathogenic |
| FAM03 (HOCM) | TNNI3 (Exon 7) | chr19:55665519; NM_000363.4 | c.428C>A | p.Thr143Asn | rs397516348 | Not reported | 0.0000362 | Not found | DM? cardiomyopathy, hypertrophic CM135615 | VUS to likely pathogenic | Conflicting interpretations of pathogenicity | Likely pathogenic |
| TTN (Exon 276) | chr2:179424125; NM_001256850.1 | c.81809_81811delAAG | p.Glu27270del | rs727504797 | Not reported | 0.000169 | Not found | Not reported | VUS to likely pathogenic | Conflicting interpretations of pathogenicity | VUS | |
| FAM04 (HCM) | FLNC (Exon 17) | chr7:128483367; NM_001458.4 | c.2635C>T | p.Arg879Cys | rs374983276 | Not reported | 0.000122 | 0.000454545 | Not reported | VUS | Conflicting interpretations of pathogenicity | VUS |
| MYPN (Exon 20) | chr10:69970208; NM_001256267.1 | c.3959T>C | p.Leu1320Pro | rs200646285 | 0.0002 | 0.00000796 | 0.001818182 | DM? noncompaction, left ventricular CM1711700 | VUS | Uncertain significance | VUS | |
| FAM05 (HCM) | MYBPC3 (Exon 33) | chr11:47353740; NM_000256.3 | c.3697C>T | p.Gln1233Ter | rs397516037 | Not reported | 0.00000802 | Not found | DM cardiomyopathy, hypertrophic CM014069 | Pathogenic | Pathogenic/ Likely pathogenic | Pathogenic |
| FAM06 (HCM) | MYH7 (Exon 14) | chr14:23898214; NM_000257.3 | c.1357C>A | p.Arg453Ser | rs121913625 | Not reported | Not reported | Not found | DM cardiomyopathy, hypertrophic CM087715 | Pathogenic | Pathogenic | Pathogenic |
| RBM20 (Exon 14) | chr10:112595636; NM_001134363.2 | c.3584C>A | p.Ser1195Tyr | rs753102653 | Not reported | 0.000278 | 0.002727273 | DM? noncompaction, left ventricular CM1711693 | VUS to likely benign | Conflicting interpretations of pathogenicity | VUS | |
| FAM07 (HCM) | RBM20 (Exon 11) | chr10:112581138; NM_001134363.2 | c.2761A>G | p.Ile921Val | rs397516608 | 0.000399 | 0.0000702 | 0.000454545 | Not reported but DM? cardiomyopathy, non-compaction with c.2761A>T, p.Ile921Phe CM1924052 | VUS | Conflicting interpretations of pathogenicity | VUS |
| FAM08 (HOCM) | RBM20 | chr10:112595737; NM_001134363.2 | c.*1T>G (in the 3′-UTR) | / | Not reported | Not reported | Not reported | Not found | Not reported | VUS to likely benign | Not reported | VUS |
| FAM09 (HOCM) | KCNQ1 (Exon 15) | chr11:2799241; NM_000218.2 | c.1768G>A | p.Ala590Thr | rs199472813 | Not reported | 0.00000797 | 0.000454545 | DM long QT syndrome CM040442 | Pathogenic | Conflicting interpretations of pathogenicity | Likely pathogenic |
| MYH7 (Exon 35) | chr14:23885010; NM_000257.3 | c.4985G>A | p.Arg1662His | rs370328209 | Not reported | 0.0000597 | 0.000454545 | DM cardiomyopathy, dilated CM115875 | VUS to likely pathogenic | Conflicting interpretations of pathogenicity | VUS | |
| DSC2 (Exon 13) | chr18:28651805; NM_024422.4 | c.1891A>G | p.Thr631Ala | Not reported | Not reported | Not reported | 0.004090909 | Not reported | VUS | Not reported | VUS | |
| FAM10 (HOCM) | PLN (Exon 2) | chr6:118880229; NM_002667.4 | c.145G>A | p.Val49Met | rs749962743 | Not reported | 0.0000119 | Not found | DM cardiomyopathy, hypertrophic CM1513486 | VUS to likely pathogenic | Uncertain significance | Likely pathogenic |
| LAMA4 | chr6:112510308; NM_001105206.2 | c.814+1_814+4delGTAA | / | rs782628388 | Not reported | 0.00002124 | 0.000909091 | Not reported | VUS to likely pathogenic | Not reported | VUS | |
| FAM11 (HCM) | MYH7 (Exon 19) | chr14:23895179; NM_000257.3 | c.2156G>A | p.Arg719Gln | rs121913641 | Not reported | Not reported | Not found | DM cardiomyopathy, hypertrophic CM941085 | Pathogenic | Pathogenic | Pathogenic |
| RYR2 (Exon 68) | chr1:237870293; NM_001035.2 | c.9625C>A | p.Pro3209Thr | rs767375014 | Not reported | 0.00000803 | 0.004090909 | Not reported but DM? sudden infant death syndrome with c.9626C>T, p.Pro3209Leu CM1824622 | VUS to likely pathogenic | Uncertain significance | VUS | |
| FAM12 (HCM) | TTN (Exon 247) | chr2:179458924; NM_001256850.1 | c.53273G>C | p.Arg17758Pro | Not reported | Not reported | Not reported | 0.001363636 | Not reported but DM? cardiomyopathy with c.53273G>A, p.Arg17758Gln CM1924179 | VUS | Not reported | VUS |
| TTN (Exon 165) | chr2:179514916; NM_001256850.1 | c.35271G>C | p.Glu11757Asp | rs1442749271 | Not reported | 0.00000465 | Not found | Not reported | VUS | Not reported | VUS | |
| FAM13 (HCM) | ALPK3 (Exon 6) | chr15:85401457; NM_020778.4 | c.4094C>T | p.Ala1365Val | rs755941827 | Not reported | 0.0000496 | 0.001818182 | Not reported | VUS to likely benign | Uncertain significance | VUS |
| FAM14 (HCM) | TTN (Exon 80) | chr2:179584550; NM_001256850.1 | c.22718G>T | p.Arg7573Ile | rs370939248 | Not reported | 0.0000124 | Not found | Not reported | VUS to likely pathogenic | Uncertain significance | VUS |
| DSP (Exon 24) | chr6:7584649; NM_004415.3 | c.7154G>A | p.Arg2385His | rs1396768987 | Not reported | 0.00000398 | Not found | Not reported | VUS to likely benign | Likely benign | VUS | |
| FAM15 (DCM) | ANK2 (Exon 43) | chr4:114290809; NM_001148.5 | c.11458C>T | p.Arg3820Trp | rs199922285 | 0.000399 | 0.0000239 | 0.009545455 | Not reported | VUS | Not reported | VUS |
| DSP (Exon 23) | chr6:7581747; NM_004415.3 | c.5324G>T | p.Arg1775Ile | rs34738426 | 0.0002 | 0.0000678 | 0.007727273 | DM cardiomyopathy, arrhythmogenic right ventricular CM056324 | VUS to likely benign | Conflicting interpretations of pathogenicity | VUS | |
| FAM16 (DCM) | LAMP2 (Exon 1) | chrX:119603022; NM_001122606.1 | c.3G>C | p.Met1Ile | Not reported | Not reported | Not reported | Not found | Not reported | Likely pathogenic | Not reported | Likely pathogenic |
| FAM17 (DCM) | NEBL (Exon 24) | chr10:21101703; NM_006393.2 | c.2513T>C | p.Ile838Thr | rs749452317 | Not reported | 0.00000398 | Not found | Not reported | VUS | Uncertain significance | VUS |
| MYL2 (Exon 6) | chr12:111350943; NM_000432.3 | c.359G>A | p.Arg120Gln | rs192057022 | 0.000399 | 0.0000517 | 0.000454545 | Not reported but DM cardiomyopathy, hypertrophic with c.358C>T, p.Arg120Trp CM1617083 | VUS to likely benign | Conflicting interpretations of pathogenicity | VUS | |
| FAM18 (DCM) | LMNA (Exon 5) | chr1:156105070; NM_170707.3 | c.908_909delCT | p.Ser303CysfsTer27 | rs59684335 | Not reported | Not reported | Not found | DM cardiomyopathy, dilated CD035724 | Pathogenic | Pathogenic | Pathogenic |
| FAM19 (DCM) | MYH7 (Exon 21) | chr14:23894624; NM_000257.3 | c.2290T>C | p.Phe764Leu | Not reported | Not reported | Not reported | Not found | Not reported but DM cardiomyopathy with c.2292C>A, p.Phe764Leu CM2037625 & DM cardiomyopathy, dilated with c.2292C>G, p.Phe764Leu CM003003 & cardiomyopathy, hypertrophic with c.2291T>A, p.Phe764Tyr CM1310641 | Pathogenic | Not reported | Pathogenic |
| MYH7 (Exon 35) | chr14:23885010; NM_000257.3 | c.4985G>A | p.Arg1662His | rs370328209 | Not reported | 0.0000597 | 0.000454545 | DM cardiomyopathy, dilated CM115875 | VUS to likely pathogenic | Conflicting interpretations of pathogenicity | VUS | |
| SCN5A (Exon 27) | chr3:38592615; NM_001160161.1 | c.5086C>T | p.Leu1696Phe | rs45606037 | Not reported | 0.00000398 | Not found | Not reported | VUS to likely pathogenic | Not reported | VUS | |
| FAM20 (DCM) | NEXN (Exon 12) | chr1:78407806; NM_144573.3 | c.1582_1584delGAA | p.Glu528del | rs764505909 | Not reported | 0.000153 | Not found | DM cardiomyopathy dilated CD1315240 | VUS to likely pathogenic | Uncertain significance | VUS |
| TTN (Exon 240) | chr2:179464037; NM_001256850.1 | c.51560A>C | p.Asn17187Thr | rs71423569 | Not reported | Not reported | 0.000909091 | Not reported | VUS | Not reported | VUS | |
| TTN (Exon 289) | chr2:179412628; NM_001256850.1 | c.88802G>A | p.Arg29601His | rs369899675 | Not reported | 0.000101 | Not found | Not reported | VUS to likely benign | Conflicting interpretations of pathogenicity | VUS | |
| FAM21 (DCM) | TTN (Exon 288) | chr2:179414482; NM_001256850.1 | c.87043_87044insCA | p.Ile29015ThrfsTer15 | Not reported | Not reported | Not reported | Not found | Not reported | Likely pathogenic | Not reported | Likely pathogenic |
| PKP2 (Exon 1) | chr12:33049482; NM_004572.3 | c.184C>A | p.Gln62Lys | rs199601548 | Not reported | 0.000141 | Not found | DM arrhythmogenic right ventricular dysplasia CM061171 | VUS | Conflicting interpretations of pathogenicity | VUS | |
| FAM22 (ARVC) | DES (Exon 1) | chr2:220283312; NM_001927.3 | c.128A>C | p.Lys43Thr | Not reported | Not reported | Not reported | Not found | Not reported but DM? cardiomyopathy, dilated with c.127A>G, p.Lys43Glu CM1616845 | VUS to likely pathogenic | Not reported | VUS |
| FAM23 (ARVC) | DSC2 (Exon 2) | chr18:28673542; NM_024422.4 | c.133delG | p.Ala45ProfsTer10 | rs1460932284 | Not reported | Not reported | Not found | DM arrhythmogenic right ventricular cardiomyopathy CD1925890 | Likely pathogenic | Not reported | Likely pathogenic |
| MYH6 (Exon 34) | chr14:23855228; NM_002471.3 | c.5072G>C | p.Arg1691Pro | Not reported | Not reported | Not reported | 0.000909091 | Not reported but DM cardiomyopathy, hypertrophic with c.5071C>T, p.Arg1691Cys CM1813213 & DM? cardiomyopathy, hypertrophic with c.5072G>A, p.Arg1691His CM204938 | VUS to likely pathogenic | Not reported | VUS | |
| FAM24 (ARVC) | DSC2 (Exon 2) | chr18:28673542; NM_024422.4 | c.133delG | p.Ala45ProfsTer10 | rs1460932284 | Not reported | Not reported | Not found | DM arrhythmogenic right ventricular cardiomyopathy CD1925890 | Likely pathogenic | Not reported | Likely pathogenic |
| DSC2 (Exon 8) | chr18:28662978; NM_024422.4 | c.991C>A | p.Gln331Lys | Not reported | Not reported | Not reported | 0.000454545 | Not reported | VUS | Not reported | Likely pathogenic | |
| FAM25 (Non-compaction cardiomyopathy) | TNNC1 (Exon 5) | chr3:52485426; NM_003280.2 | c.435C>A | p.Asp145Glu | rs267607124 | Not reported | 0.000132 | Not found | DM cardiomyopathy, hypertrophic CM083569 | Likely pathogenic | Uncertain significance | VUS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koutsofti, C.; Ioannides, M.; Polydorou, C.; Papagregoriou, G.; Malatras, A.; Michael, G.; Hadjiioannou, I.; Pieri, S.; Loizidou, E.M.; Eftychiou, C.; et al. Massive Parallel DNA Sequencing of Patients with Inherited Cardiomyopathies in Cyprus and Suggestion of Digenic or Oligogenic Inheritance. Genes 2024, 15, 319. https://doi.org/10.3390/genes15030319
Koutsofti C, Ioannides M, Polydorou C, Papagregoriou G, Malatras A, Michael G, Hadjiioannou I, Pieri S, Loizidou EM, Eftychiou C, et al. Massive Parallel DNA Sequencing of Patients with Inherited Cardiomyopathies in Cyprus and Suggestion of Digenic or Oligogenic Inheritance. Genes. 2024; 15(3):319. https://doi.org/10.3390/genes15030319
Chicago/Turabian StyleKoutsofti, Constantina, Marios Ioannides, Christiana Polydorou, Gregory Papagregoriou, Apostolos Malatras, George Michael, Irene Hadjiioannou, Stylianos Pieri, Eleni M. Loizidou, Christos Eftychiou, and et al. 2024. "Massive Parallel DNA Sequencing of Patients with Inherited Cardiomyopathies in Cyprus and Suggestion of Digenic or Oligogenic Inheritance" Genes 15, no. 3: 319. https://doi.org/10.3390/genes15030319
APA StyleKoutsofti, C., Ioannides, M., Polydorou, C., Papagregoriou, G., Malatras, A., Michael, G., Hadjiioannou, I., Pieri, S., Loizidou, E. M., Eftychiou, C., Papasavvas, E., Christophides, T., Alkelai, A., Kapoor, M., Shuldiner, A. R., Avraamides, P., & Deltas, C. (2024). Massive Parallel DNA Sequencing of Patients with Inherited Cardiomyopathies in Cyprus and Suggestion of Digenic or Oligogenic Inheritance. Genes, 15(3), 319. https://doi.org/10.3390/genes15030319