

Translation of Data from Animal Models of Cancer to Immunotherapy of Breast Cancer and Chronic Lymphocytic Leukemia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Use of Xenotransplant Animal Models and In Vitro Studies in CLL (See Refs. [15,23] for Details)
2.1. Methodology … CLL Studies
2.1.1. Human Plasma and Engraftment of Human CLL in NOD-SCIDγcnull Mice
2.1.2. CD200 Blockade and T-Cell Depletion In Vivo Studies
2.1.3. FACS Analyses
2.1.4. Cell Culture and Transwell Assay [19]
2.2. Results from In Vivo Studies
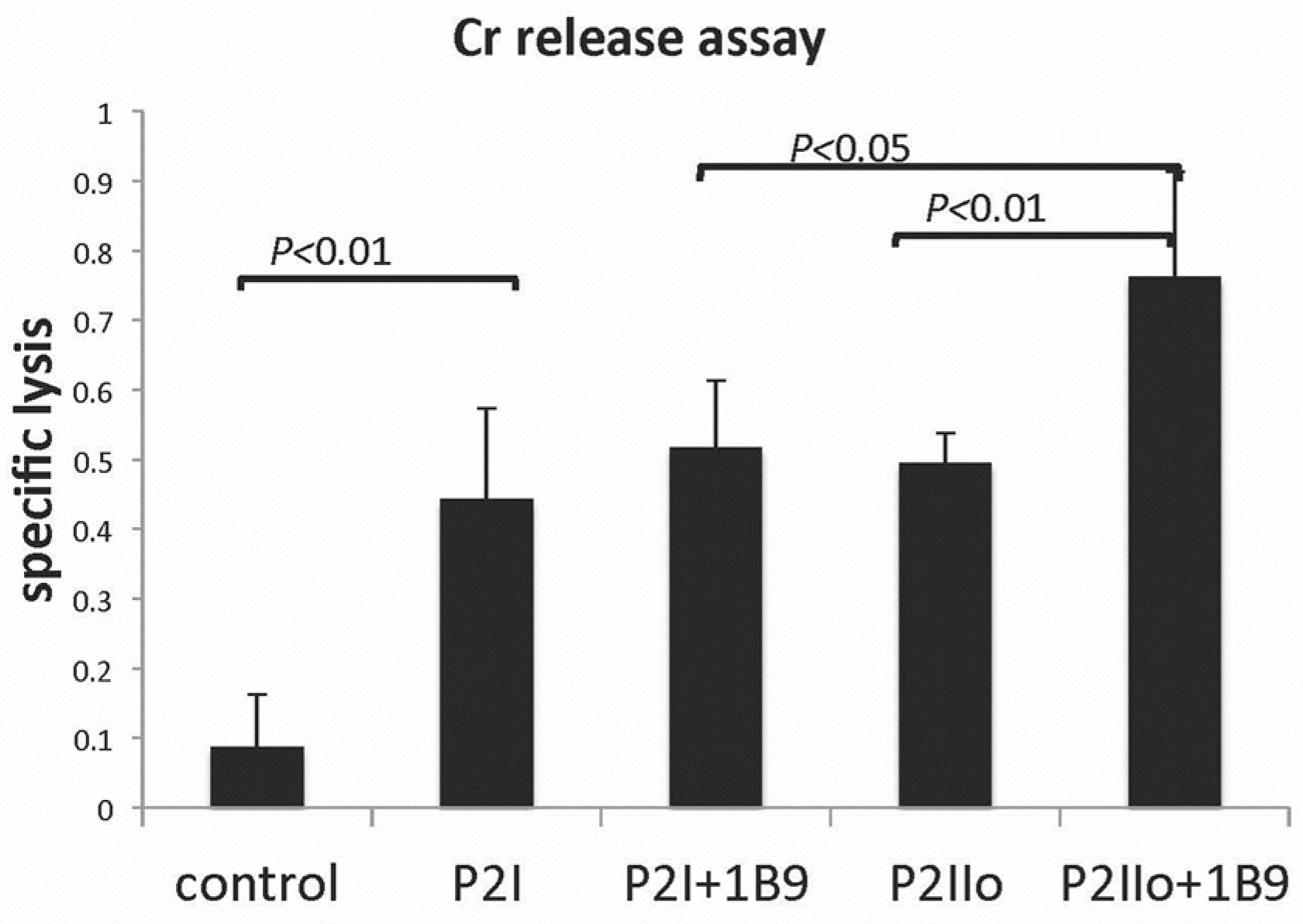
2.3. Results from In Vitro Studies
2.4. A Model System Exploring Modified CLL Cells as a Tumor Vaccine
2.5. Supportive Data for CLL Studies in Animal Models in the Literature
3. Use of Animal Models and In Vitro Studies in Breast Cancer (See Refs. [14,20] for Details)
3.1. Methodology… Breast Cancer Studies
3.1.1. 3D Culture System to Assess Tumor Invasion In Vitro
3.1.2. ELISA Assay for TNFa, IL-6, IL-8, and IL-17 In Vivo and in 3D Cultures [20]
3.2. Results from In Vivo Studies
3.3. A Model System Exploring Breast Cancer Cells as a Tumor Vaccine
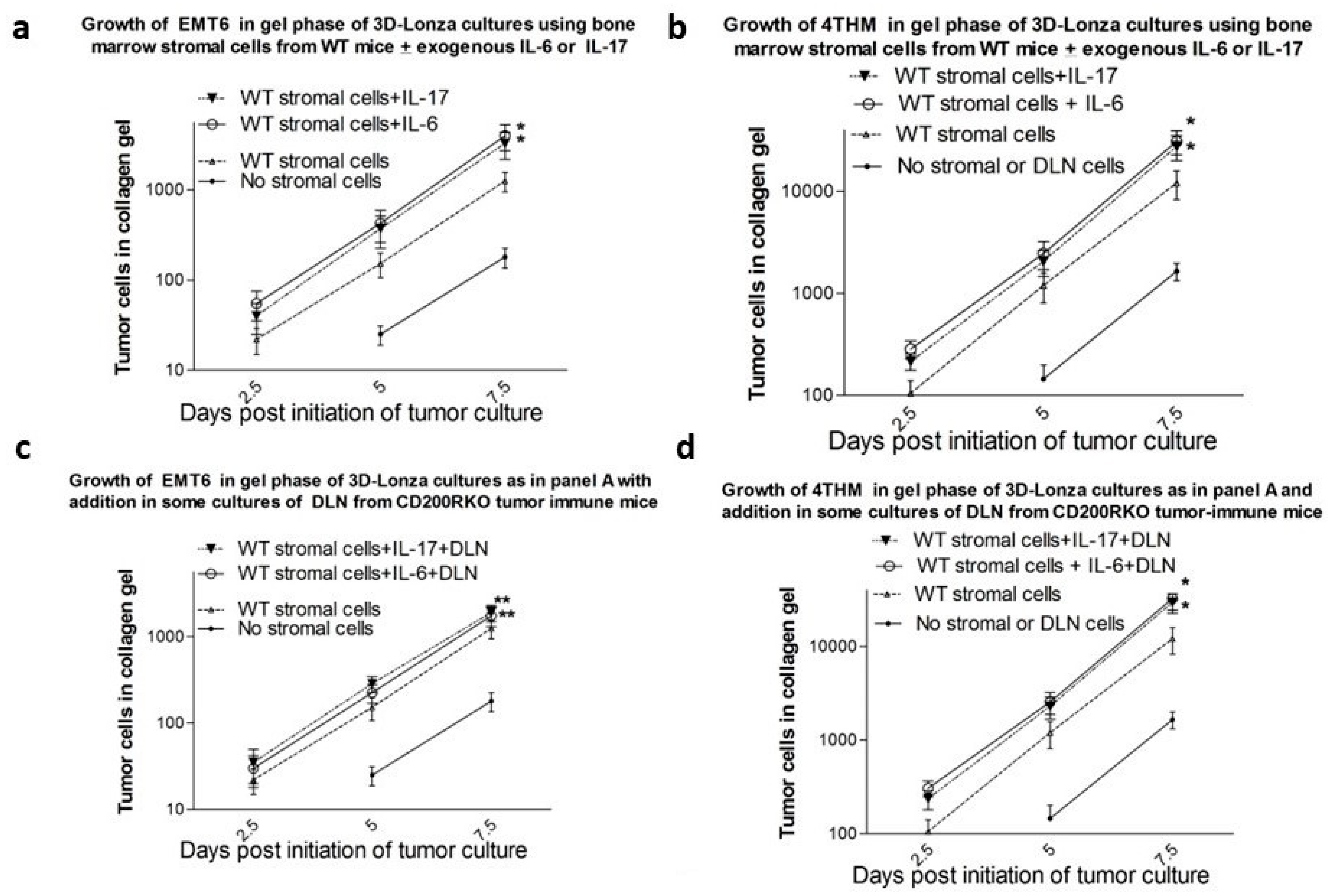
3.4. Results from In Vitro Studies
3.5. Supportive and Cautionary Data for Breast Cancer Studies in Animal Models from the Literature, including Other Solid Tumor Models
4. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adams, J.L.; Duffy, K.J.; Moore, M.L.; Yang, J. Cancer Immunotherapy—An Emerging Field That Bridges Oncology and Immunology Research; Chackalamannil, S., Rotella, D., Ward, S., Eds.; Comprehensive Medicinal Chemistry III; Elsevier: Amsterdam, The Netherlands, 2017; pp. 357–394. ISBN 9780128032015. [Google Scholar] [CrossRef]
- Ansari, M.J.; Salama, A.D.; Chitnis, T.; Smith, R.N.; Yagita, H.; Akiba, H.; Yamazaki, T.; Azuma, M.; Iwai, H.; Khoury, S.J.; et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J. Exp. Med. 2003, 198, 63–69. [Google Scholar] [CrossRef]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef]
- Chambers, C.A.; Sullivan, T.J.; Allison, J.P. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity 1997, 7, 885–895. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, M.; Jiang, W.; Wang, L.; Fu, Z.; Li, D.; Pang, D. B7-H4 gene polymorphisms are associated with sporadic breast cancer in a Chinese Han population. BMC Cancer 2009, 9, 394. [Google Scholar] [CrossRef]
- Lines, J.L.; Pantazi, E.; Mak, J.; Sempere, L.F.; Wang, L.; O’Connell, S.; Cerraz, S.; Suriawintia, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfoeld, E.A.; Coyle, A.J.; Sobel, R.A. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Okazaki, I.M.; Wang, J.; Sugira, D.; Nakaki, F.; Yoshida, T.; Kato, Y.; Fagarasan, S.; Muramatsu, M.; Eto, T. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med. 2011, 208, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Korman, A.J.; Peggs, K.S.; Allison, J.P. Checkpoint blockade in cancer immunotherapy. Adv. Immunol. 2006, 90, 297–339. [Google Scholar] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Dong, C. New checkpoints in cancer immunotherapy. Immunol. Rev. 2017, 276, 52–65. [Google Scholar] [CrossRef]
- Gorczynski, R.M. Updates on the Importance of CD200:CD200R Checkpoint Blockade in Solid Tumors and B cell Malignancies. J. Oncol. Res. Ther. 2023, 8, 10185. [Google Scholar] [CrossRef]
- Dagar, G.; Gupta, A.; Masoodi, T.; Nisar, S.; Merhi, M.; Hashem, S.; Chauhan, R.; Dagar, M.; Mirza, S.; Bagga, P.; et al. Harnessing the potential of CAR-T cell therapy: Progress, challenges, and future directions in hematological and solid tumor treatments. J. Transl. Med. 2023, 21, 449, Erratum in J. Transl. Med. 2023, 21, 571. [Google Scholar] [CrossRef]
- Gorczynski, R.; Chen, Z.; Erin, N.; Khatri, I.; Podnos, A. Comparison of immunity in mice cured of primary/metastatic growth of EMT6 or 4THM breast cancer by chemotherapy or immunotherapy. PLoS ONE 2014, 9, e113597. [Google Scholar] [CrossRef]
- Zhu, F.; Khatri, I.; Spaner, D.E.; Gorczynski, R.M. An autologous tumor vaccine for CLL. Leuk. Res. 2018, 68, 40–47. [Google Scholar] [CrossRef]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Tiwari, A.; Trivedi, R.; Lin, S.Y. Tumor microenvironment: Barrier or opportunity towards effective cancer therapy. J. Biomed. Sci. 2022, 29, 83. [Google Scholar] [CrossRef]
- Gorczynski, R.M. IL-17 in the tumor microenvironment. Adv. Exp. Med. Biol. 2020, 1240, 47–58. [Google Scholar] [CrossRef]
- Zhu, F.; McCaw, L.; Spaner, D.E.; Gorczynski, R.M. Targeting the IL-17/IL-6 axis can alter growth of Chronic Lymphocytic Leukemian in vivo/in vitro. Leuk. Res. 2018, 66, 28–38. [Google Scholar] [CrossRef]
- Gorczynski, R.M.; Erin, N.; Maqbool, T.; Gorczynski, C.P.; Gorczynski, L.Y. Characterization of an in vitro model system to explore control of tumor invasion of EMT6 and 4THM breast tumors by CD200:CD200R interactions. Breast Cancer 2018, 25, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Y.; Lin, K.; Wang, X.; Tu, Y.; Zhuo, Z. Progress in building clinically relevant patient-derived tumor xenograft models for cancer research. Anim. Model. Exp. Med. 2023, 6, 381–398. [Google Scholar] [CrossRef] [PubMed]
- Roarty, K.; Echeverria, G.V. Laboratory Models for Investigating Breast Cancer Therapy Resistance and Metastasis. Front. Oncol. 2021, 11, 645698. [Google Scholar] [CrossRef]
- Wong, K.K.; Bannerman, F.; Chesny, A.; Spaner, D.E.; Gorczynski, R.M. Soluble CD200 Is Critical to Engraft Chronic Lymphocytic Leukemia Cells in Immunocompromised Mice. Cancer Res. 2012, 72, 4931. [Google Scholar] [CrossRef]
- Soma, L.A.; Craig, F.E.; Swerdlow, S.H. The proliferation center microenvironment and prognostic markers in chronic lymphocytic leukemia/small lymphocytic lymphoma. Hum. Pathol. 2006, 37, 152–159. [Google Scholar] [CrossRef]
- Ghia, P.; Circosta, P.; Scielzo, C.; Vallario, A.; Camporeale, A.; Granziero, L.; Caligaris-Cappio, F. Differential effects on CLL cell survival exerted by different microenvironmental elements. Curr. Top. Microbiol. Immunol. 2005, 294, 135–145. [Google Scholar] [PubMed]
- Ding, W.; Nowakowski, G.S.; Knox, T.R.; Boysen, J.C.; Maas, M.L.; Schwager, S.M.; Wu, W.; Wellik, L.E.; Dietz, A.B.; Ghosh, A.K.; et al. Bi-directional activation between mesenchymal stem cells and CLL B-cells: Implication for CLL disease progression. Br. J. Haematol. 2009, 147, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Gorgun, G.; Holderried, T.A.; Zahrieh, D.; Neuberg, D.; Gribben, J.G. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J. Clin. Investig. 2005, 115, 1797–1805. [Google Scholar] [CrossRef]
- Wong, K.K.; Khatri, I.; Shaha, S.; Spaner, D.E.; Gorczynski, R.M. The role of CD200 in immunity to B cell lymphoma. J. Leukoc. Biol. 2010, 88, 361–372. [Google Scholar] [CrossRef]
- Moreaux, J.; Hose, D.; Reme, T.; Jourdan, E.; Hundemer, M.; Legouffe, E.; Moine, P.; Bourin, P.; Moos, M.; Corre, J.; et al. CD200 is a new prognostic factor in multiple myeloma. Blood 2006, 108, 4194–4197. [Google Scholar] [CrossRef]
- Petermann, K.B.; Rozenberg, G.I.; Zedek, D.; Groben, G.; McKinnon, K.; Buehler, C.; Kim, W.Y.; Shields, J.M.; Penland, S.; Bear, J.E.; et al. CD200 is induced by ERK and is a potential therapeutic target in melanoma. J. Clin. Investig. 2007, 117, 3922–3929. [Google Scholar] [CrossRef]
- Tonks, A.; Hills, R.; White, P.; Rosie, B.; I Mills, K.; Burnett, A.K.; Darley, R.L. CD200 as a prognostic factor in acute myeloid leukaemia. Leukemia 2007, 21, 566–568. [Google Scholar] [CrossRef]
- Ohishi, M.; Schipani, E. Bone marrow mesenchymal stem cells. J. Cell. Biochem. 2010, 109, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; O’Brien, S.; Maushouri, T.; Rogers, A.; Kantarjian, H.; Keating, M.; Albitar, M. Prognostic value of plasma interleukin-6 levels in patients with chronic lymphocytic leukemia. Cancer 2002, 95, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Wierda, W.G.; Johnson, M.M.; Do, K.A.; Manshouri, T.; Dey, A.; O’Brien, S.; Giles, F.J.; Kantarjian, H.; Thomas, D.; Faderl, S.; et al. Plasma interleukin 8 level predicts for survival in chronic lymphocytic leukaemia. Br. J. Haematol. 2003, 120, 452–456. [Google Scholar] [CrossRef]
- Wang, L.; Yi, T.; Kortylewski, M.; Pardoll, D.M.; Zeng, D.; Yu, H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J. Exp. Med. 2009, 206, 1457–1464. [Google Scholar] [CrossRef]
- Jin, W.; Dong, C. IL-17 cytokines in immunity and inflammation. Emerg. Microbes Infec. 2013, 2, e60. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Wang, S.; Cai, J.; Shi, J.; Sui, X.; Cao, Y.; Huang, W.; Chen, X.; Cai, Z.; et al. Bone marrow-derived mesenchymal stem cell-secreted IL-8 promotes the angiogenesis and growth of colorectal cancer. Oncotarget 2015, 6, 42825–42837. [Google Scholar] [CrossRef]
- Yan, X.J.; Dozmorov, I.; Li, W.; Yancopoulos, S.; Sison, C.; Centola, M.; Jain, P.; Allen, S.L.; Kolitz, J.E.; Rai, K.R.; et al. Identification of outcome-correlated cytokine clusters in chronic lymphocytic leukemia. Blood 2011, 118, 5201–5210. [Google Scholar] [CrossRef]
- Djouad, F.; Plence, P.; Bony, C.; Tropel, P.; Apparailly, F.; Sany, J.; Noel, D.; Jorgensen, C. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood 2003, 102, 3837–3844. [Google Scholar] [CrossRef]
- Pallasch, C.P.; Ulbrich, S.; Brinker, R.; Hallek, M.; Uger, R.A.; Wendtner, C.M. Disruption of T cell suppression in chronic lymphocytic leukemia by CD200 blockade. Leuk. Res. 2009, 33, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, J.W.; Shi, M.; Horna, P.; Olteanu, H. Increased CD200 expression in post-transplant lymphoproliferative disorders correlates with an increased frequency of FoxP3(+) regulatory T cells. Ann. Diagn. Pathol. 2020, 48, 151585. [Google Scholar] [CrossRef]
- Cieniewicz, B.; Uyeda, M.J.; Chen, P.P.; Sayitoglu, E.C.; Liu, J.M.; Andolfi, G.; Greenthal, K.; Bertaina, A.; Gregori, S.; Bacchetta, R.; et al. Engineered type 1 regulatory T cells designed for clinical use kill primary pediatric acute myeloid leukemia cells. Haematologica 2021, 106, 2588–2597. [Google Scholar] [CrossRef]
- Long, M.; Beckwith, K.; Do, P.; Mundy, B.L.; Gordon, A.; Lehman, A.M.; Maddocks, K.J.; Cheney, C.; Jones, J.A.; Flynn, J.M.; et al. Ibrutinib treatment improves T cell number and function in CLL patients. J. Clin. Investig. 2017, 127, 3052–3064. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Hu, A.; Zhu, J.; Yu, J.; Talebian, F.; Bai, X.F. CD200-CD200R Pathway in the Regulation of Tumor Immune Microenvironment and Immunotherapy. Adv. Exp. Med. Biol. 2020, 1223, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Okuma, K. Mouse Models for HTLV-1 Infection and Adult T Cell Leukemia. Int. J. Mol. Sci. 2023, 24, 11737. [Google Scholar] [CrossRef]
- Xin, Q.; Chen, Z.; Wei, W.; Wu, Y. Animal models of acute lymphoblastic leukemia: Recapitulating the human disease to evaluate drug efficacy and discover therapeutic targets. Biochem. Pharmacol. 2022, 198, 114970. [Google Scholar] [CrossRef]
- Alexander, T.C.; Krull, K.R. Effects of chemotherapy for acute lymphoblastic leukemia on cognitive function in animal models of contemporary protocols: A systematic literature review. Neurosci. Biobehav. Rev. 2021, 129, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Konantz, M.; Schürch, C.; Hanns, P.; Müller, J.S.; Sauteur, L.; Lengerke, C. Modeling hematopoietic disorders in zebrafish. Dis. Model. Mech. 2019, 12, dmm040360. [Google Scholar] [CrossRef] [PubMed]
- Ten Hacken, E.; Wu, C.J. Understanding CLL biology through mouse models of human genetics. Blood 2021, 138, 2621–2631. [Google Scholar] [CrossRef]
- Pekarsky, Y.; Zanesi, N.; Aqeilan, R.I.; Croce, C.M. Animal models for chronic lymphocytic leukemia. J. Cell. Biochem. 2007, 100, 1109–1118. [Google Scholar] [CrossRef]
- Erin, N.; Podnos, A.; Tanriover, G.; Duymuş, O.; Cote, E.; Khatri, I.; Gorczynski, R.M. Bidirectional effect of CD200 on breast cancer development and metastasis, with ultimate outcome determined by tumor aggressiveness and a cancer-induced inflammatory response. Oncogene 2015, 34, 3860–3870. [Google Scholar] [CrossRef]
- Podnos, A.; Clark, D.A.; Erin, N.; Yu, K.; Gorczynski, R.M. Further evidence for a role of tumor CD200 expression in breast cancer tumor invasion: Decreased tumor invasion in CD200R1KO mice or using CD200-silenced EMT6. Breast Cancer Res. Treat. 2012, 136, 117–127. [Google Scholar] [CrossRef]
- Curry, A.; Khatri, I.; Kos, O.; Zhu, F.; Gorczynski, R. Importance of CD200 expression by tumor or host cells to regulation of immunotherapy in a mouse breast cancer model. PLoS ONE 2017, 12, e0171586. [Google Scholar] [CrossRef]
- Nip, C.; Wang, L.; Liu, C. CD200/CD200R: Bidirectional Role in Cancer Progression and Immunotherapy. Biomedicines 2023, 11, 3326. [Google Scholar] [CrossRef] [PubMed]
- Gorczynski, R.M.; Erin, N.; Zhu, F. Serum-derived exosomes from mice with highly metastatic breast cancer transfer increased metastatic capacity to a poorly metastatic tumor. Cancer Med. 2016, 5, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Gorczynski, R.M.; Podnos, A.; Kos, O.; Oyedele, A.; Zhu, F.; Khatri, I. Significance of Soluble CD200 in plasma and serum of human breast cancer patients. Int. Med. Rev. 2016, 2. [Google Scholar] [CrossRef]
- Fang, J.; Chen, F.; Liu, D.; Gu, F.; Chen, Z.; Wang, Y. Prognostic value of immune checkpoint molecules in breast cancer. Biosci. Rep. 2020, 40, BSR20201054. [Google Scholar] [CrossRef] [PubMed]
- Magdeldin, T.; López-Dávila, V.; Villemant, C.; Cameron, G.; Drake, R.; Cheema, U.; Loizidou, M. The efficacy of cetuximab in a tissue-engineered three-dimensional in vitro model of colorectal cancer. J. Tissue Eng. 2014, 5, 2041731414544183. [Google Scholar] [CrossRef]
- Zhao, P.; Chen, Y.; Yue, Z.; Yuan, Y.; Wang, X. Bone marrow mesenchymal stem cells regulate stemness of multiple myeloma cell lines via BTK signaling pathway. Leuk. Res. 2017, 57, 20–26. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Glentis, A.; Oertle, P.; Mariani, P.; Chikina, A.; El Marjou, F.; Attieh, Y.; Zaccarini, F.; Lae, M.; Loew, D.; Dingli, F.; et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat. Commun. 2017, 8, 924. [Google Scholar] [CrossRef]
- Jin, K.; Pandey, N.B.; Popel, A.S. Crosstalk between stromal components and tumor cells of TNBC via secreted factors enhances tumor growth and tumor invasion. Oncotarget 2017, 8, 60210–60222. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and tumor invasion. Nat. Cell Biol. 2014, 16, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Pandit, T.S.; Kennette, W.; MacKenzie, L. Lymphatic metastasis of breast cancer cells is associated with differential gene expression profiles that predict cancer stem cell- like properties and the ability to survive, establish and grow in a foreign environment. Int. J. Oncol. 2009, 35, 297–308. [Google Scholar] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, H.-W.; Lu, M.-H.; He, X.-H.; Li, Y.; Gu, H.; Liu, M.-F.; Wang, E.-D. MicroRNA-155 Functions as an OncomiR in Breast Cancer by Targeting the Suppressor of Cytokine Signaling 1 Gene. Cancer Res. 2010, 70, 3119–3127. [Google Scholar] [CrossRef]
- Yang, F.; Ning, Z.; Ma, L.; Liu, W.; Shao, C.; Shu, Y.; Shen, H. Exosomal miRNAs and miRNA dysregulation in cancer-associated fibroblasts. Oncotarget 2017, 8, 21609–21625. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Tumor invasion. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and tumor invasion of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef]
- Chen, Y.; Zeng, C.; Zhan, Y.; Wang, H.; Jiang, X.; Li, W. Aberrant low expression of p85α in stromal fibroblasts promotes breast cancer cell tumor invasion through exosome-mediated paracrine Wnt10b. Oncogene 2017, 36, 4692–4705. [Google Scholar] [CrossRef]
- Cioffi, M.; Trabulo, S.M.; Vallespinos, M.; Raj, D.; Kheir, T.B.; Lin, M.L.; Begum, J.; Baker, A.M.; Amgheib, A.; Saif, J.; et al. The miR-25-93-106b cluster regulates tumor tumor invasion and immune evasion via modulation of CXCL12 and PD-L1. Mol. Cancer 2017, 16, 148. [Google Scholar] [CrossRef]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32, 303–331. [Google Scholar] [CrossRef] [PubMed]
- Shao, A.; Owens, D.M. The immunoregulatory protein CD200 as a potentially lucrative yet elusive target for cancer therapy. Oncotarget 2023, 14, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, W.; Zhang, S. Seeking for correlative genes and signaling pathways with bone metastasis from breast cancer by integrated analysis. Front. Oncol. 2019, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Vathiotis, I.A.; MacNeil, T.; Zugazagoitia, J.; Syrigos, K.N.; Aung, T.N.; Gruver, A.M.; Vaillancourt, P.; Hughes, I.; Hinton, S.; Driscoll, K.; et al. Quantitative assessment of CD200 and CD200R expression in lung cancer. Cancers 2021, 13, 1024. [Google Scholar] [CrossRef]
- Sun, H.; Xu, J.; Huang, M.; Huang, Q.; Sun, R.; Xiao, W.; Sun, C. CD200R, a co-inhibitory receptor on immune cells, predicts the prognosis of human hepatocellular carcinoma. Immunol. Lett. 2016, 178, 105–113. [Google Scholar] [CrossRef]
- Klatka, J.; Grywalska, E.; Klatka, M.; Wasiak, M.; Andrzejczak, A.; Rolinski, J. Expression of selected regulatory molecules on the CD83+ monocyte-derived dendritic cells generated from patients with laryngeal cancer and their clinical significance. Eur. Arch. Otorhinolaryngol. 2013, 270, 2683–2693. [Google Scholar] [CrossRef]
- Choueiry, F.; Torok, M.; Shakya, R.; Agrawal, K.; Deems, A.; Benner, B.; Hinton, A.; Shaffer, J.; Blaser, B.W.; Noonan, A.M.; et al. CD200 promotes immunosuppression in the pancreatic tumor microenvironment. J. Immunother. Cancer 2020, 8, e000189. [Google Scholar] [CrossRef]
- Tronik-Le Roux, D.; Sautreuil, M.; Bentriou, M.; Verine, J.; Palma, M.B.; Daouya, M.; Bouhidel, F.; Lemler, S.; LeMaoult, J.; Desgrandchamps, F.; et al. Comprehensive landscape of immune-checkpoints uncovered in clear cell renal cell carcinoma reveals new and emerging therapeutic targets. Cancer Immunol. Immunother. 2020, 69, 1237–1252. [Google Scholar] [CrossRef]
- Khan, I.Z.; Del Guzzo, C.A.; Shao, A.; Cho, J.; Du, R.; Cohen, A.O.; Owens, D.M. The CD200-CD200R axis promotes squamous cell carcinoma metastasis via regulation of cathepsin k. Cancer Res. 2021, 81, 5021–5032. [Google Scholar] [CrossRef]
- Gaiser, M.R.; Weis, C.A.; Gaiser, T.; Jiang, H.; Buder-Bakhaya, K.; Herpel, E.; Warth, A.; Xiao, Y.; Miao, L.; Brownell, I. Merkel cell carcinoma expresses the immunoregulatory ligand CD200 and induces immunosuppressive macrophages and regulatory t cells. Oncoimmunology 2018, 7, e1426517. [Google Scholar] [CrossRef]
- Moertel, C.L.; Xia, J.; LaRue, R.; Waldron, N.N.; Andersen, B.M.; Prins, R.M.; Okada, H.; Donson, A.M.; Foreman, N.K.; Hunt, M.A.; et al. CD200 in cns tumor-induced immunosuppression: The role for CD200 pathway blockade in targeted immunotherapy. J. Immunother. Cancer. 2014, 2, 46. [Google Scholar] [CrossRef]
- Xin, C.; Zhu, J.; Gu, S.; Yin, M.; Ma, J.; Pan, C.; Tang, J.; Zhang, P.; Liu, Y.; Bai, X.F.; et al. CD200 is overexpressed in neuroblastoma and regulates tumor immune microenvironment. Cancer Immunol. Immunother. 2020, 69, 2333–2343. [Google Scholar] [CrossRef]
- Mihrshahi, R.; Brown, M.H. Downstream of tyrosine kinase 1 and 2 play opposing roles in CD200 receptor signaling. J. Immunol. 2010, 185, 7216–7222. [Google Scholar] [CrossRef]
- Lin, C.H.; Talebian, F.; Li, Y.; Zhu, J.; Liu, J.Q.; Zhao, B.; Basu, S.; Pan, X.; Chen, X.; Yan, P.; et al. CD200R signaling contributes to unfavorable tumor microenvironment through regulating production of chemokines by tumor-associated myeloid cells. iScience 2023, 26, 106904. [Google Scholar] [CrossRef]
- Fraser, S.D.; Sadofsky, L.R.; Kaye, P.M.; Hart, S.P. Reduced expression of monocyte CD200R is associated with enhanced proinflammatory cytokine production in sarcoidosis. Sci. Rep. 2016, 6, 38689. [Google Scholar] [CrossRef]
- Stumpfova, M.; Ratner, D.; Desciak, E.B.; Eliezri, Y.D.; Owens, D.M. The immunosuppressive surface ligand CD200 augments the metastatic capacity of squamous cell carcinoma. Cancer Res. 2010, 70, 2962–2972. [Google Scholar] [CrossRef]
- Liao, K.L.; Bai, X.F.; Friedman, A. The role of CD200-CD200R in tumor immune evasion. J. Theor. Biol. 2013, 328, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Ampudia-Mesias, E.; Shaver, R.; Horbinski, C.M.; Moertel, C.L.; Olin, M.R. Tumor-derived vaccines containing CD200 inhibit immune activation: Implications for immunotherapy. Immunotherapy 2016, 8, 1059–1071. [Google Scholar] [CrossRef]
- Olin, M.R.; Ampudia-Mesias, E.; Pennell, C.A.; Sarver, A.; Chen, C.C.; Moertel, C.L.; Hunt, M.A.; Pluhar, G.E. Treatment Combining CD200 Immune Checkpoint Inhibitor and Tumor-Lysate Vaccination after Surgery for Pet Dogs with High-Grade Glioma. Cancers 2019, 11, 137. [Google Scholar] [CrossRef]
- Ampudia-Mesias, E.; Puerta-Martinez, F.; Bridges, M.; Zellmer, D.; Janeiro, A.; Strokes, M.; Sham, Y.Y.; Taher, A.; Castro, M.G.; Moertel, C.L.; et al. CD200 Immune-Checkpoint Peptide Elicits an Anti-glioma Response Through the DAP10 Signaling Pathway. Neurotherapeutics 2021, 18, 1980–1994, Erratum in: Neurotherapeutics 2021, 18, 213. [Google Scholar] [CrossRef] [PubMed]
- Gorczynski, R.M.; Kai, Y.; Lee, L.; Chen, Z.; Clark, D.A.; Wong, S.; Marsden, P.A. Structural and functional heterogeneity in the CD200R family of immunoregulatory molecules. Am. J. Reprod. Immunol. 2004, 52, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Pilch, Z.; Tonecka, K.; Skorzynski, M.; Sas, Z.; Braniewska, A.; Kryczka, T.; Boon, L.; Golab, J.; Meyaard, L.; Rygiel, T.P. The pro-tumor effect of CD200 expression is not mimicked by agonistic CD200R antibodies. PLoS ONE 2019, 14, e0210796. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Yano, H.; Umakoshi, A.; Matsumoto, S.; Mise, A.; Funahashi, Y.; Ueno, Y.; Kamei, Y.; Takada, Y.; Kumon, Y.; et al. A Truncated form of CD200 (CD200S) Expressed on Glioma Cells Prolonged Survival in a Rat Glioma Model by Induction of a Dendritic Cell-Like Phenotype in Tumor-Associated Macrophages. Neoplasia 2016, 18, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, D.X.; Kai, Y.; Khatri, I.; Lamptey, B.; Gorczynski, R.M. Identification of an expressed truncated form of CD200, CD200tr, which is a physiologic antagonist of CD200-induced suppression. Transplantation 2009, 86, 1116–1297. [Google Scholar] [CrossRef]
- Shin, S.P.; Goh, A.R.; Ju, J.M.; Kang, H.G.; Kim, S.J.; Kim, J.K.; Park, E.J.; Bae, Y.S.; Choi, K.; Jung, Y.S.; et al. Local adenoviral delivery of soluble CD200R-ig enhances antitumor immunity by inhibiting CD200-β-catenin-driven m2 macrophage. Mol. Ther. Oncolytics 2021, 23, 138–150. [Google Scholar] [CrossRef]
- Talebian, F.; Yu, J.; Lynch, K.; Liu, J.Q.; Carson, W.E.; Bai, X.F. CD200 Blockade Modulates Tumor Immune Microenvironment but Fails to Show Efficacy in Inhibiting Tumor Growth in a Murine Model of Melanoma. Front. Cell Dev. Biol. 2021, 9, 739816. [Google Scholar] [CrossRef]
- Yi, M.; Zheng, X.; Niu, M.; Zhu, S.; Ge, H.; Wu, K. Combination strategies with PD-1/PD-L1 blockade: Current advances and future directions. Mol. Cancer 2022, 21, 28. [Google Scholar] [CrossRef]
- Ge, Y.; Xi, H.; Ju, S.; Zhang, X. Blockade of PD-1/PD-L1 immune checkpoint during DC vaccination induces potent protective immunity against breast cancer in hu-SCID mice. Cancer Lett. 2013, 336, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Fellermeyer, M.; Anzilotti, C.; Paluch, C.; Cornall, R.J.; Davis, S.J.; Gileadi, U. Combination CD200R/PD-1 blockade in a humanised mouse model. Immunother. Adv. 2023, 3, ltad006. [Google Scholar] [CrossRef]
- Mou, J.; Zheng, W.; Wei, D.; Li, D.; Fan, R.; Tang, Q. CD200-CD200R affects cisplatin and paclitaxel sensitivity by regulating cathepsin K-mediated p65 NF-κB signaling in cervical cancer. Heliyon 2023, 9, e19220. [Google Scholar] [CrossRef]
- Moon, S.Y.; Han, M.; Ryu, G.; Shin, S.A.; Lee, J.H.; Lee, C.S. Emerging Immune Checkpoint Molecules on Cancer Cells: CD24 and CD200. Int. J. Mol. Sci. 2023, 24, 15072. [Google Scholar] [CrossRef]
- Yoshimura, K.; Suzuki, Y.; Inoue, Y.; Tsuchiya, K.; Karayama, M.; Iwashita, Y.; Kahyo, T.; Kawase, A.; Tanahashi, M.; Ogawa, H.; et al. CD200 and CD200R1 are differentially expressed and have differential prognostic roles in non-small cell lung cancer. Oncoimmunology 2020, 9, 1746554. [Google Scholar] [CrossRef]
- Bisgin, A.; Meng, W.J.; Adell, G.; Sun, X.F. Interaction of CD200 Overexpression on Tumor Cells with CD200R1 Overexpression on Stromal Cells: An Escape from the Host Immune Response in Rectal Cancer Patients. J. Oncol. 2019, 2019, 5689464. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Duan, L.; Li, H.; Liu, X.; Cai, T.; Yang, Y.; Yin, Y.; Chang, W.; Zhong, L.; Zhang, L.; et al. PD1hi CD200hi CD4+ exhausted T cell increase immunotherapy resistance and tumour progression by promoting epithelial-mesenchymal transition in bladder cancer. Clin. Transl. Med. 2023, 13, e1303. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, T.; Zheng, L.; Liu, H.; Song, W.; Liu, D.; Li, Z.; Pan, C.X. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 156. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorczynski, R. Translation of Data from Animal Models of Cancer to Immunotherapy of Breast Cancer and Chronic Lymphocytic Leukemia. Genes 2024, 15, 292. https://doi.org/10.3390/genes15030292
Gorczynski R. Translation of Data from Animal Models of Cancer to Immunotherapy of Breast Cancer and Chronic Lymphocytic Leukemia. Genes. 2024; 15(3):292. https://doi.org/10.3390/genes15030292
Chicago/Turabian StyleGorczynski, Reginald. 2024. "Translation of Data from Animal Models of Cancer to Immunotherapy of Breast Cancer and Chronic Lymphocytic Leukemia" Genes 15, no. 3: 292. https://doi.org/10.3390/genes15030292
APA StyleGorczynski, R. (2024). Translation of Data from Animal Models of Cancer to Immunotherapy of Breast Cancer and Chronic Lymphocytic Leukemia. Genes, 15(3), 292. https://doi.org/10.3390/genes15030292