
Homozygous CNP Mutation and Neurodegeneration in Weimaraners: Myelin Abnormalities and Accumulation of Lipofuscin-like Inclusions
 , ,
, ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
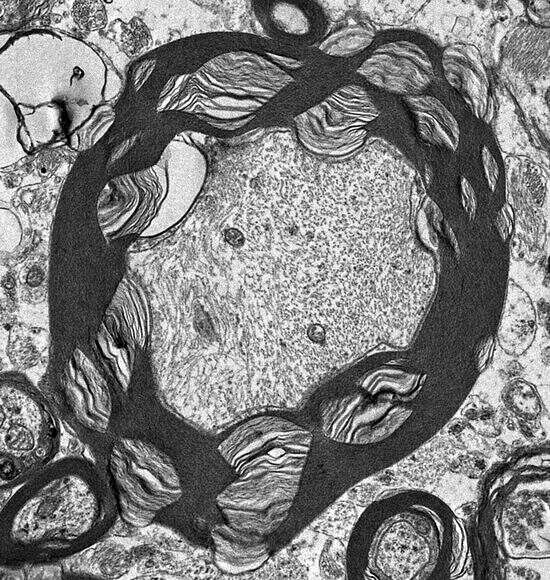
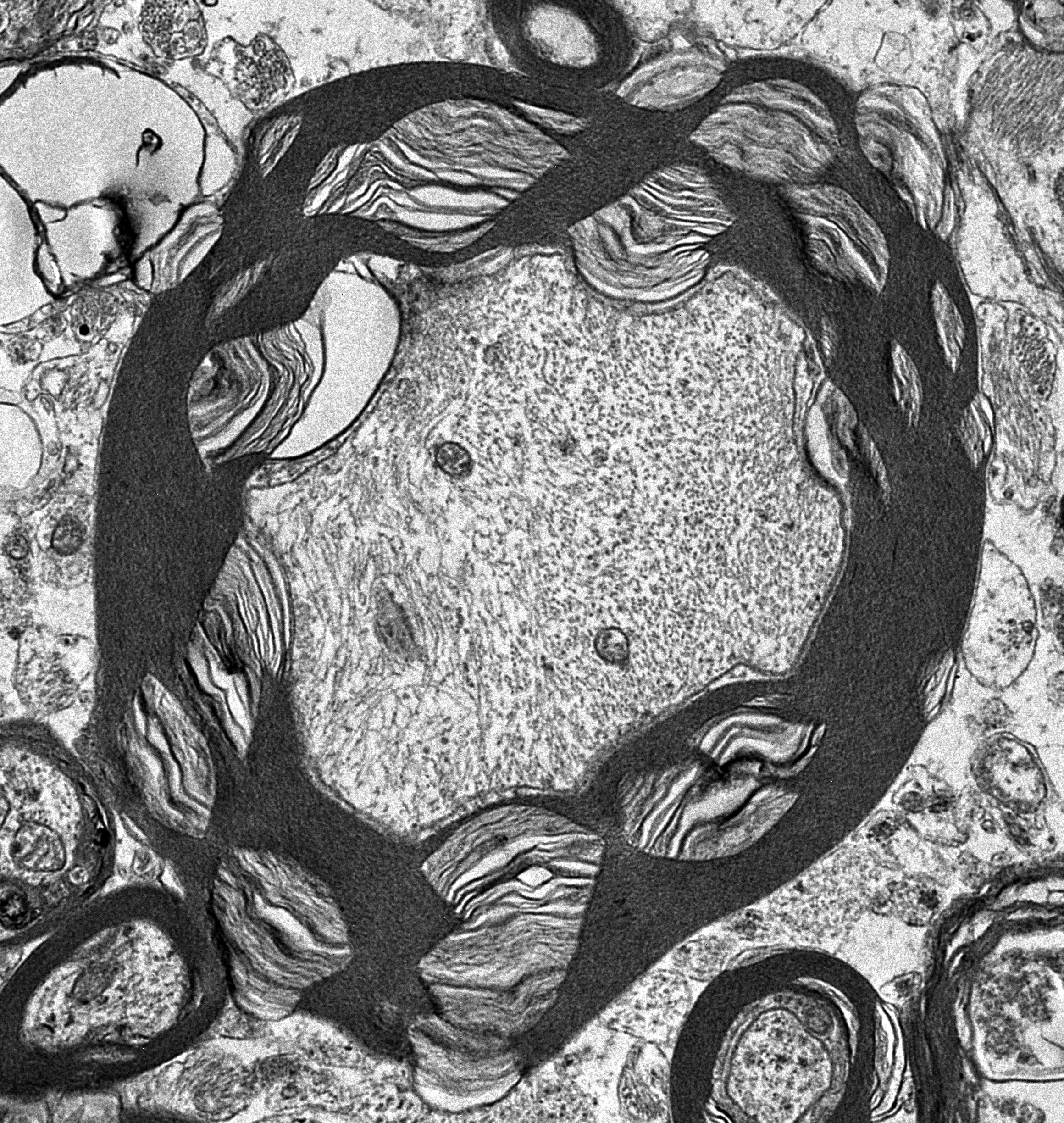
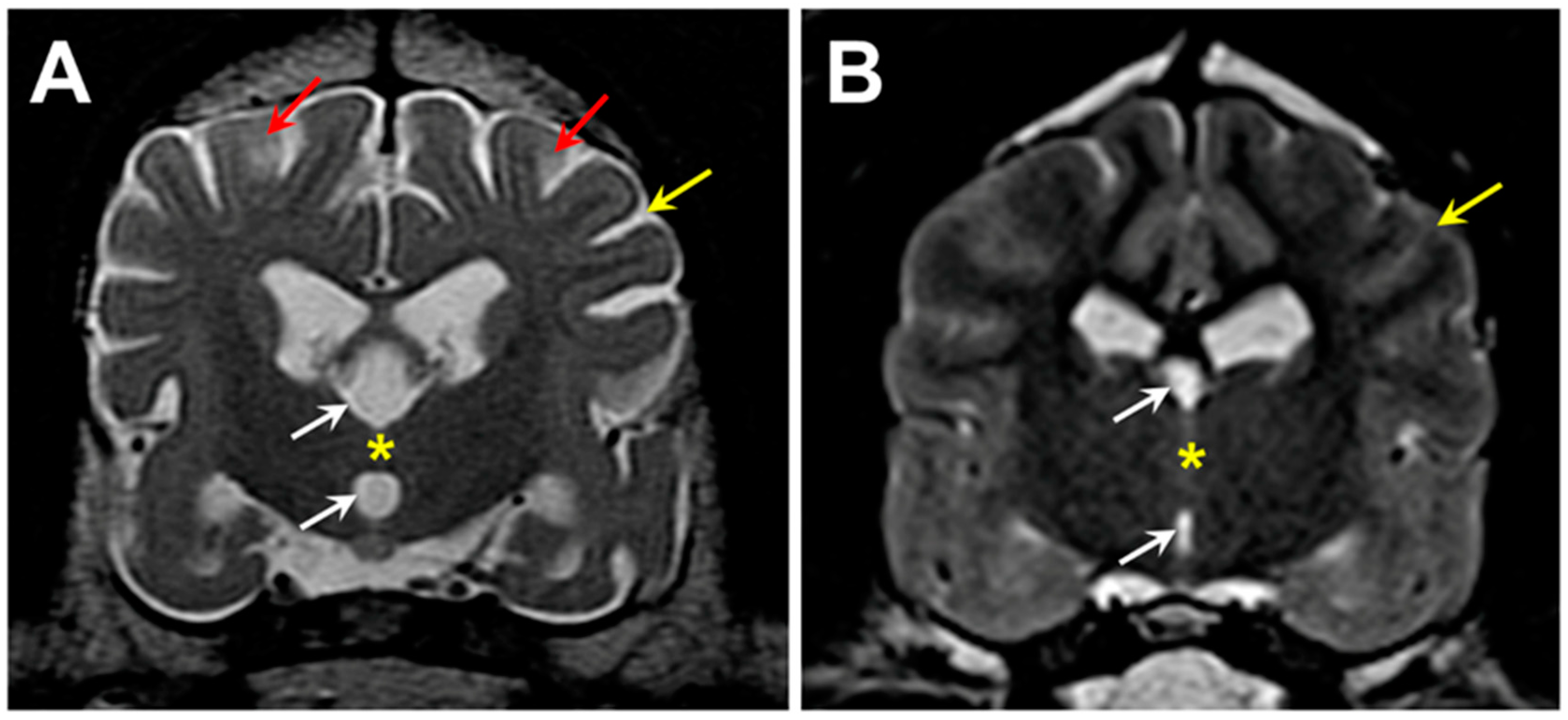
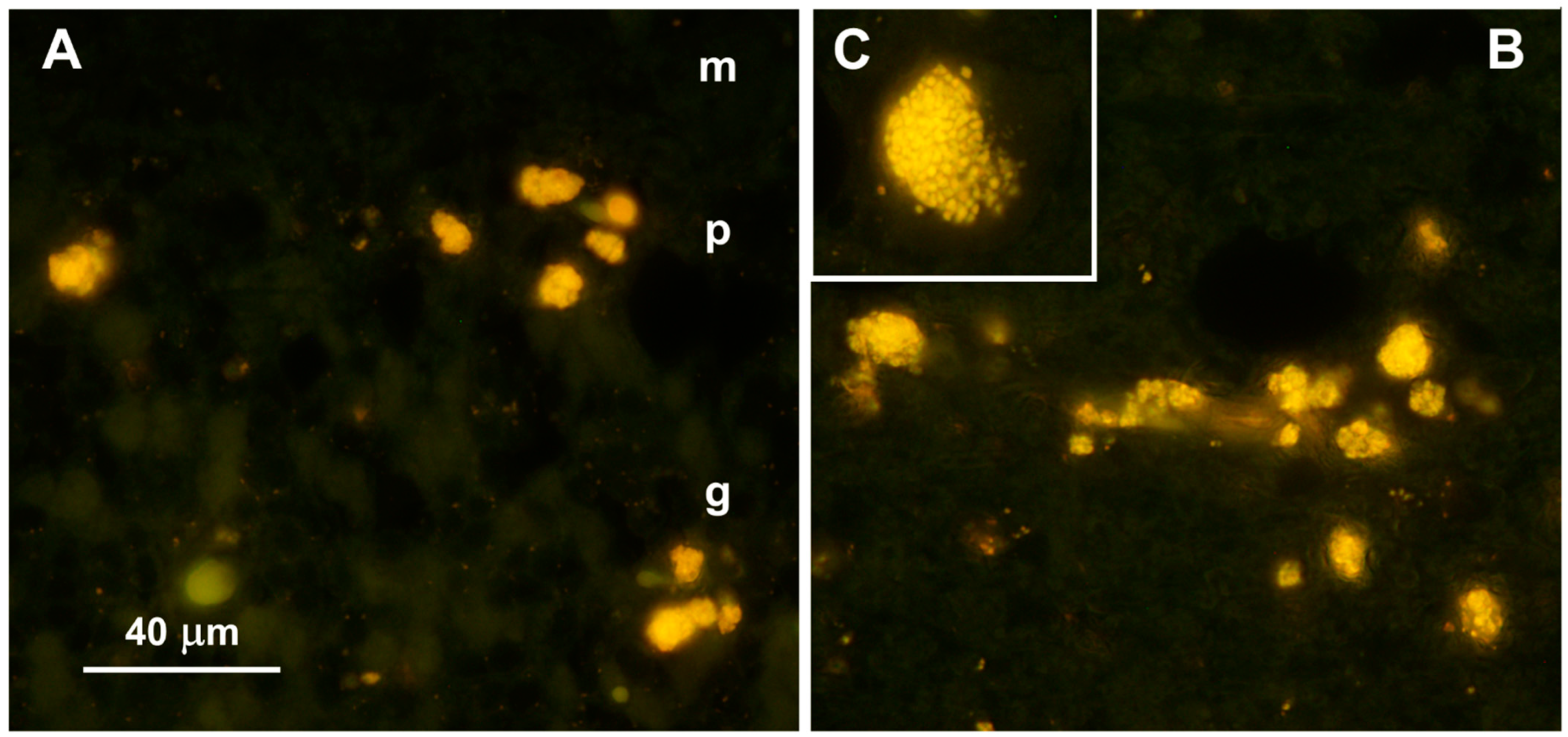
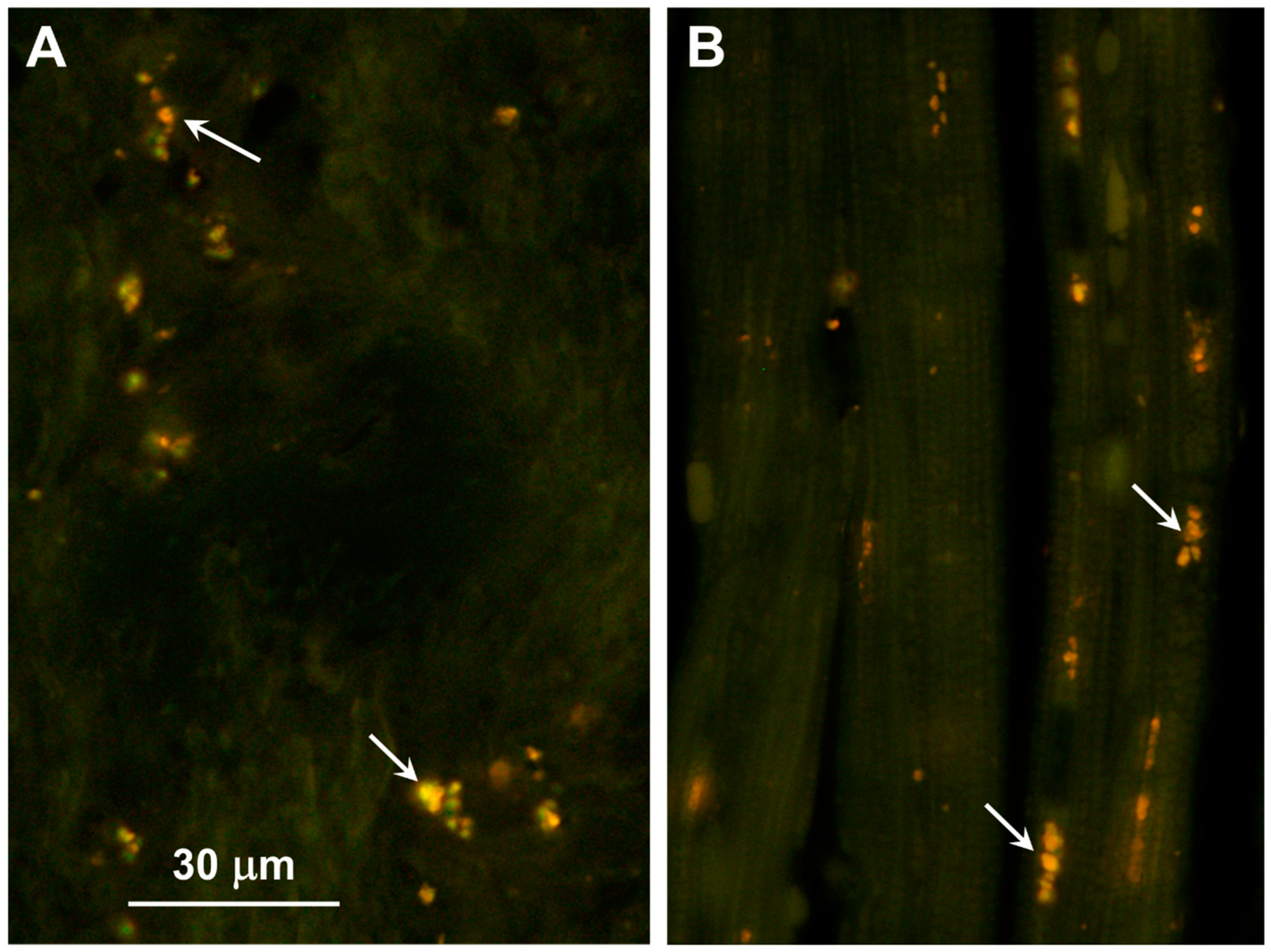
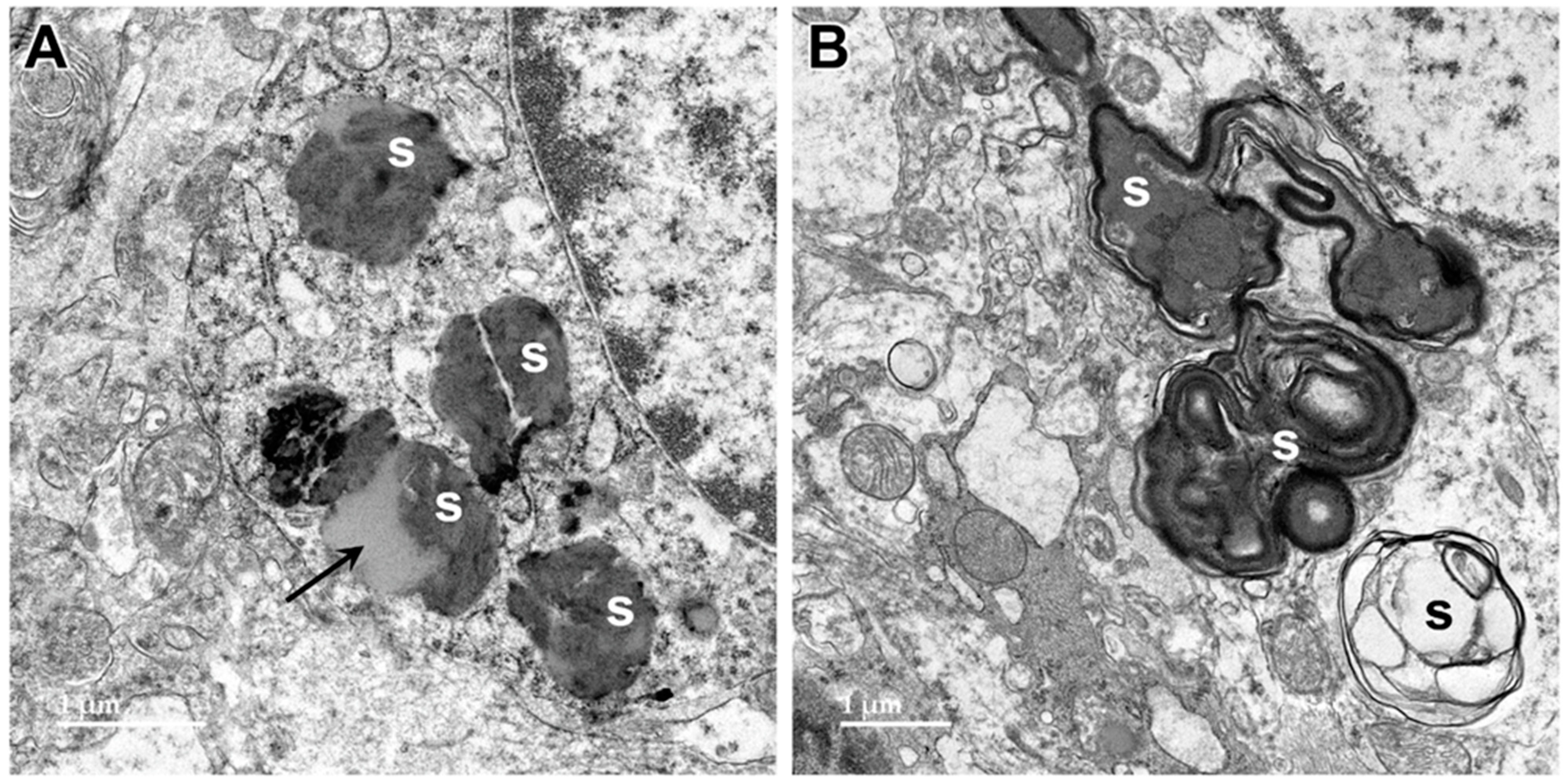
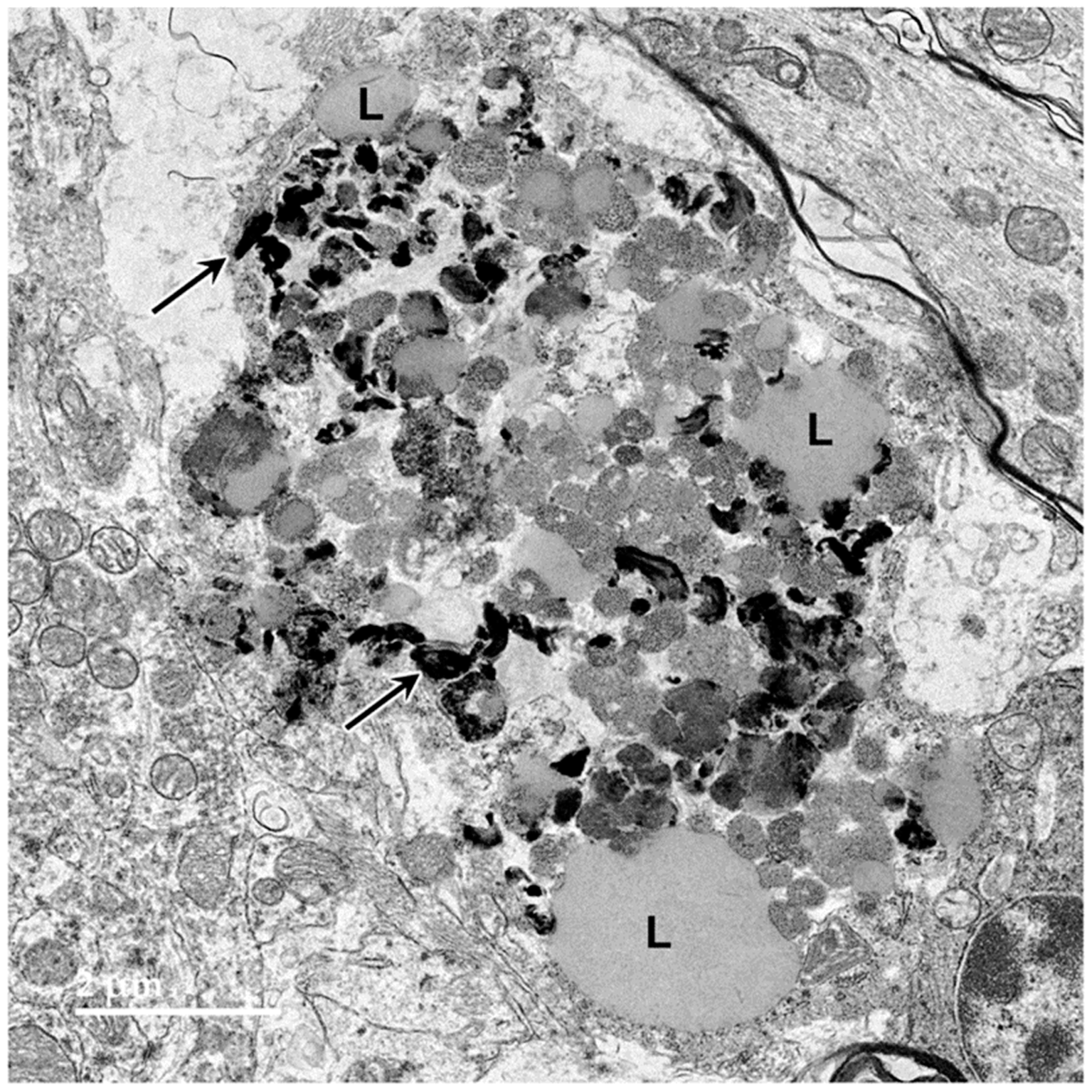
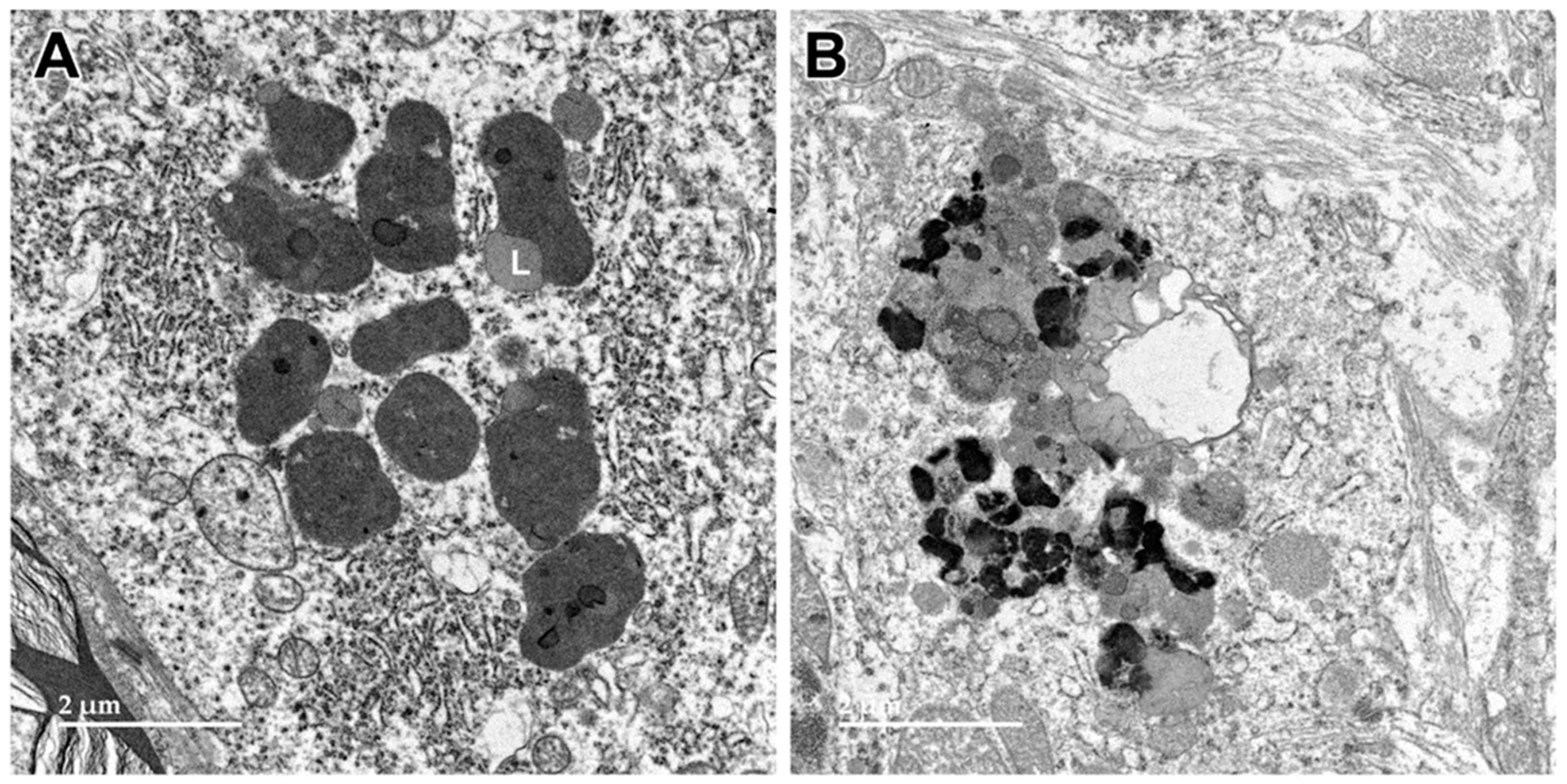
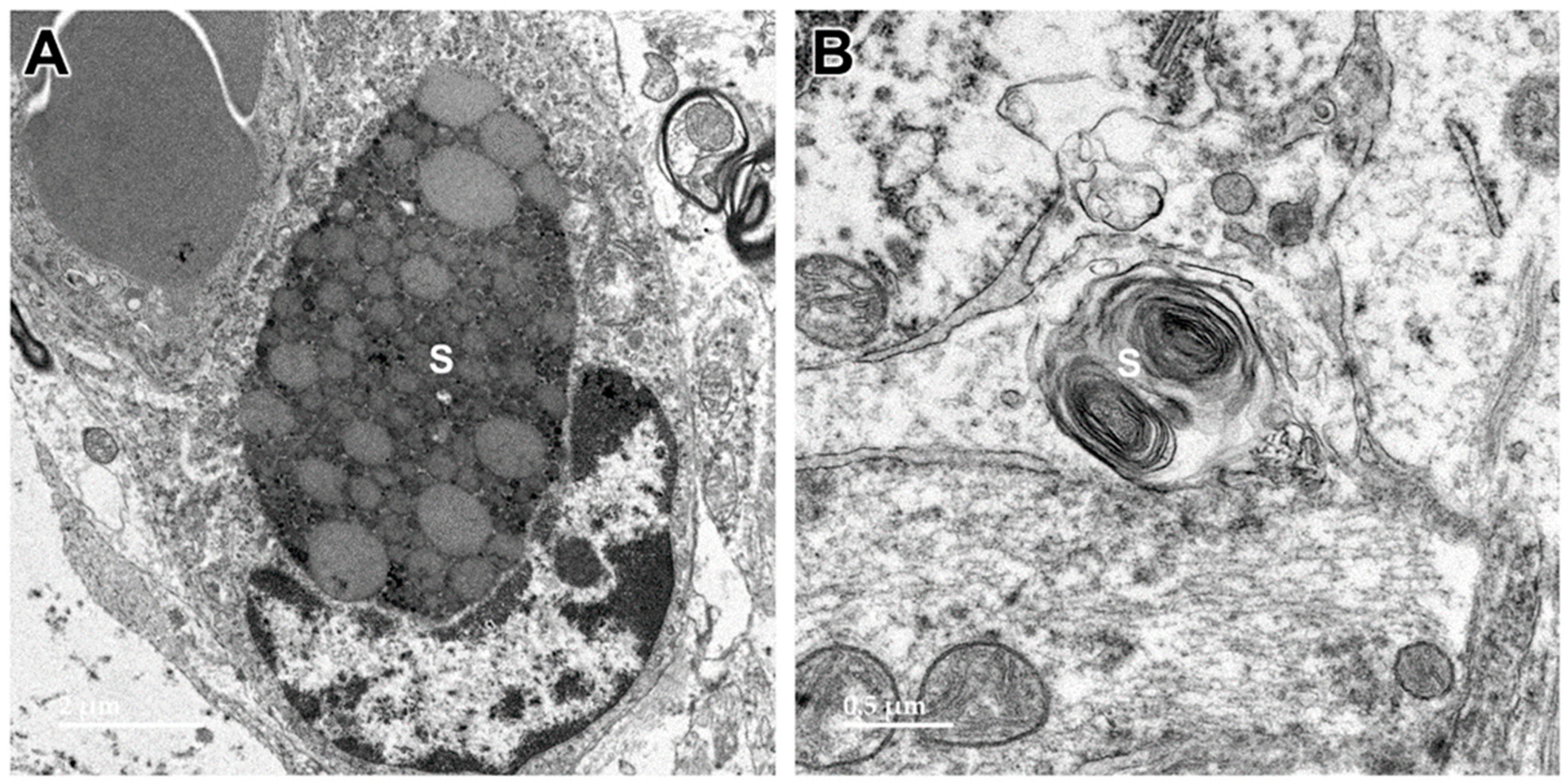
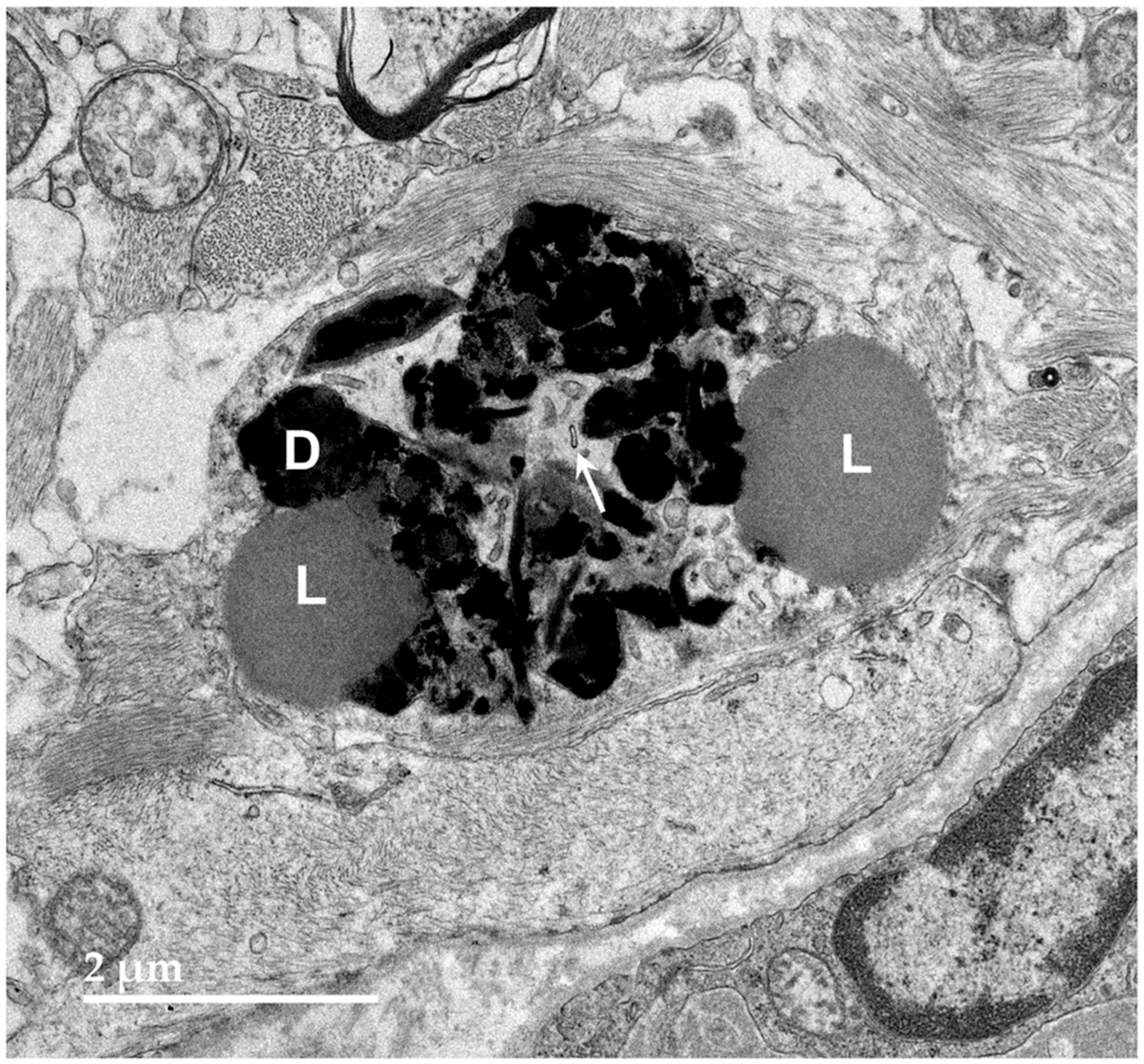
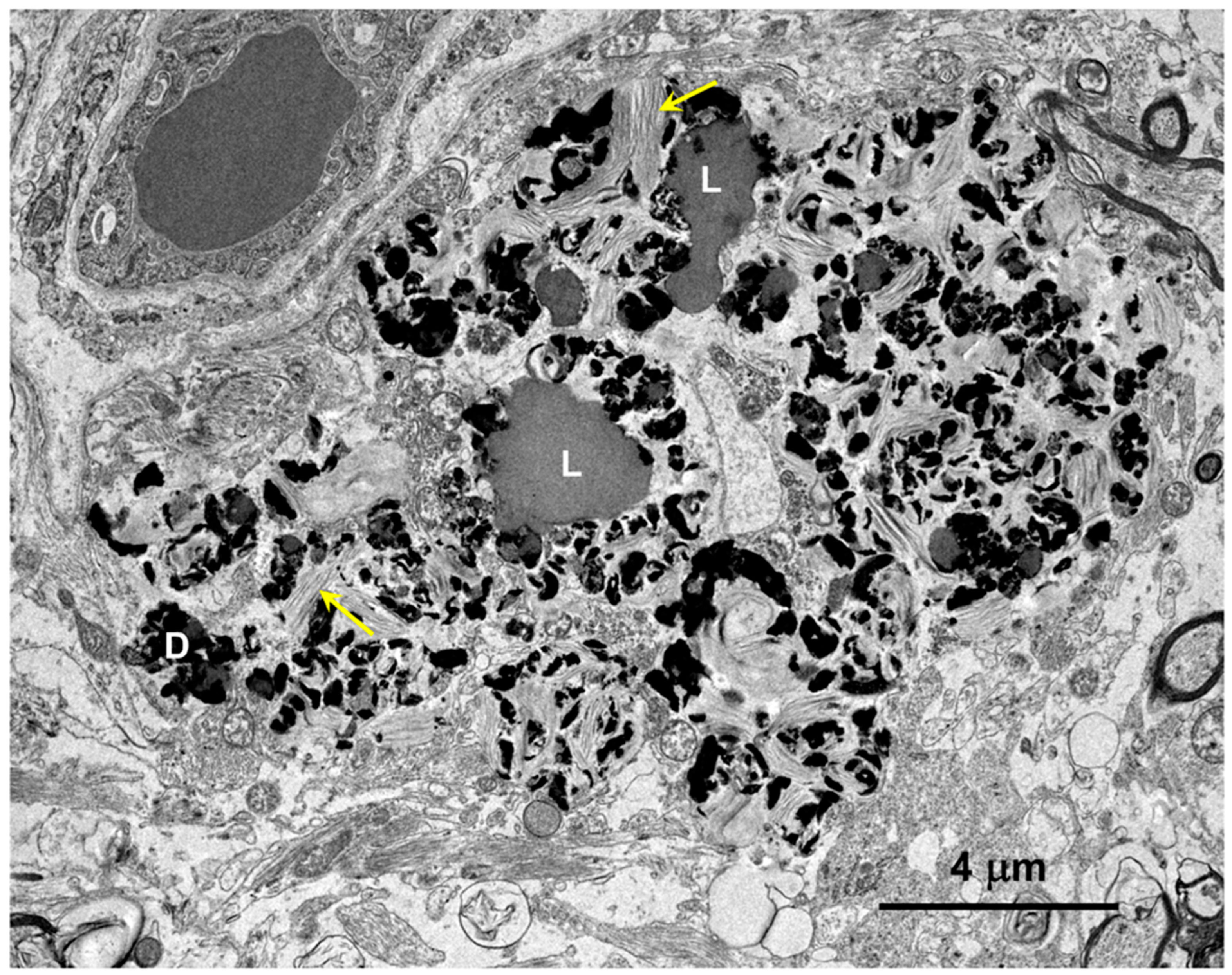
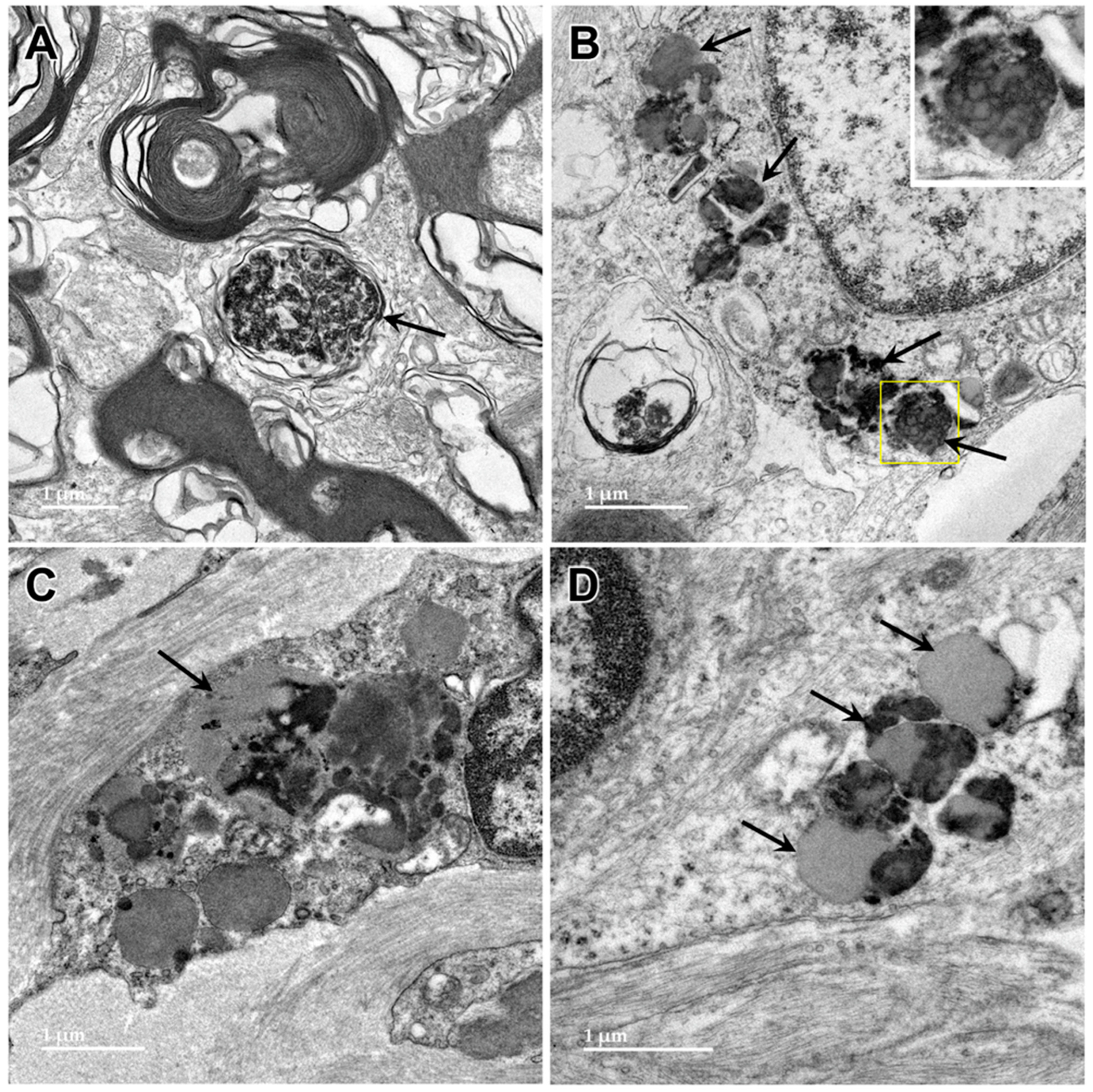
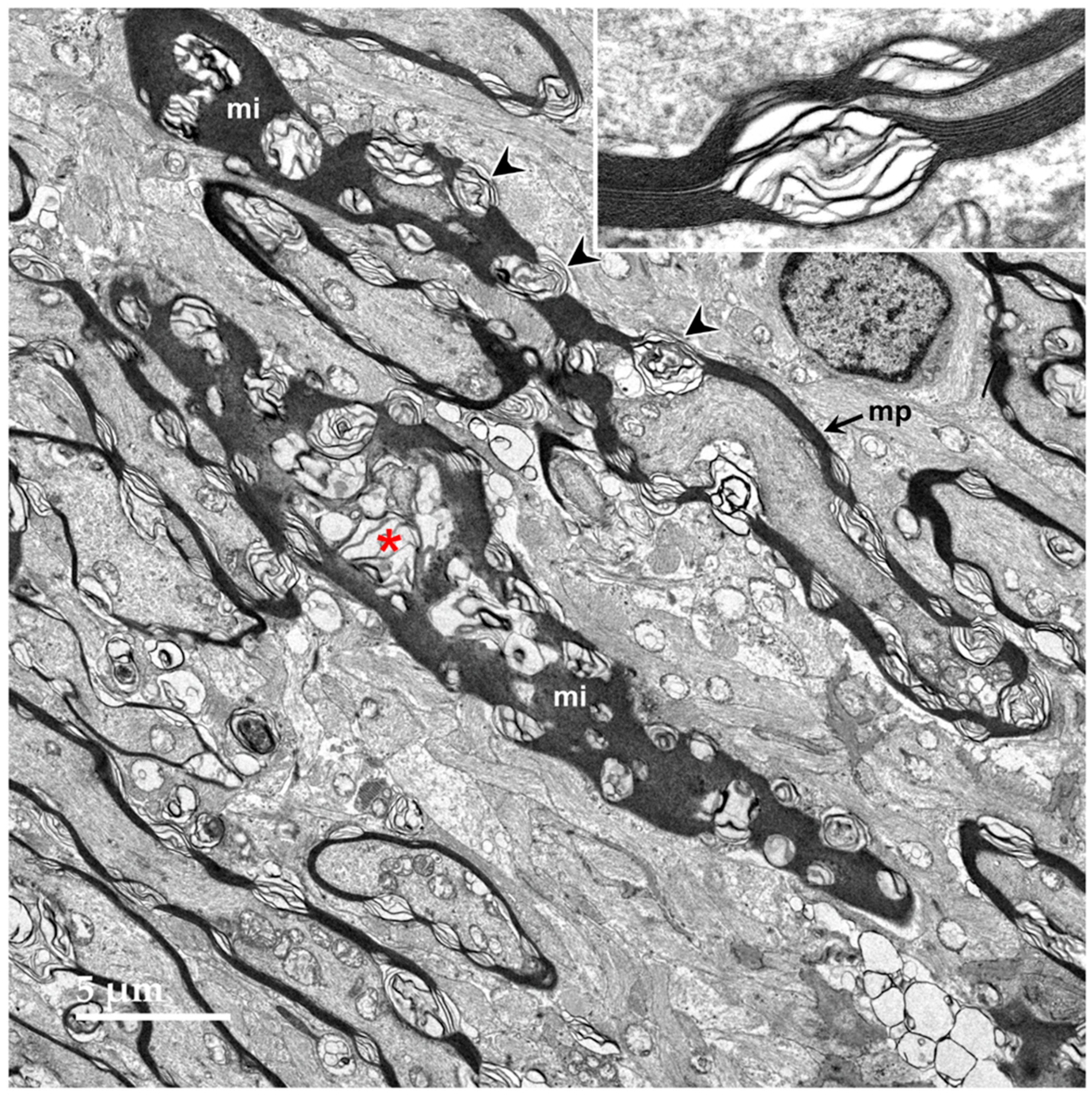
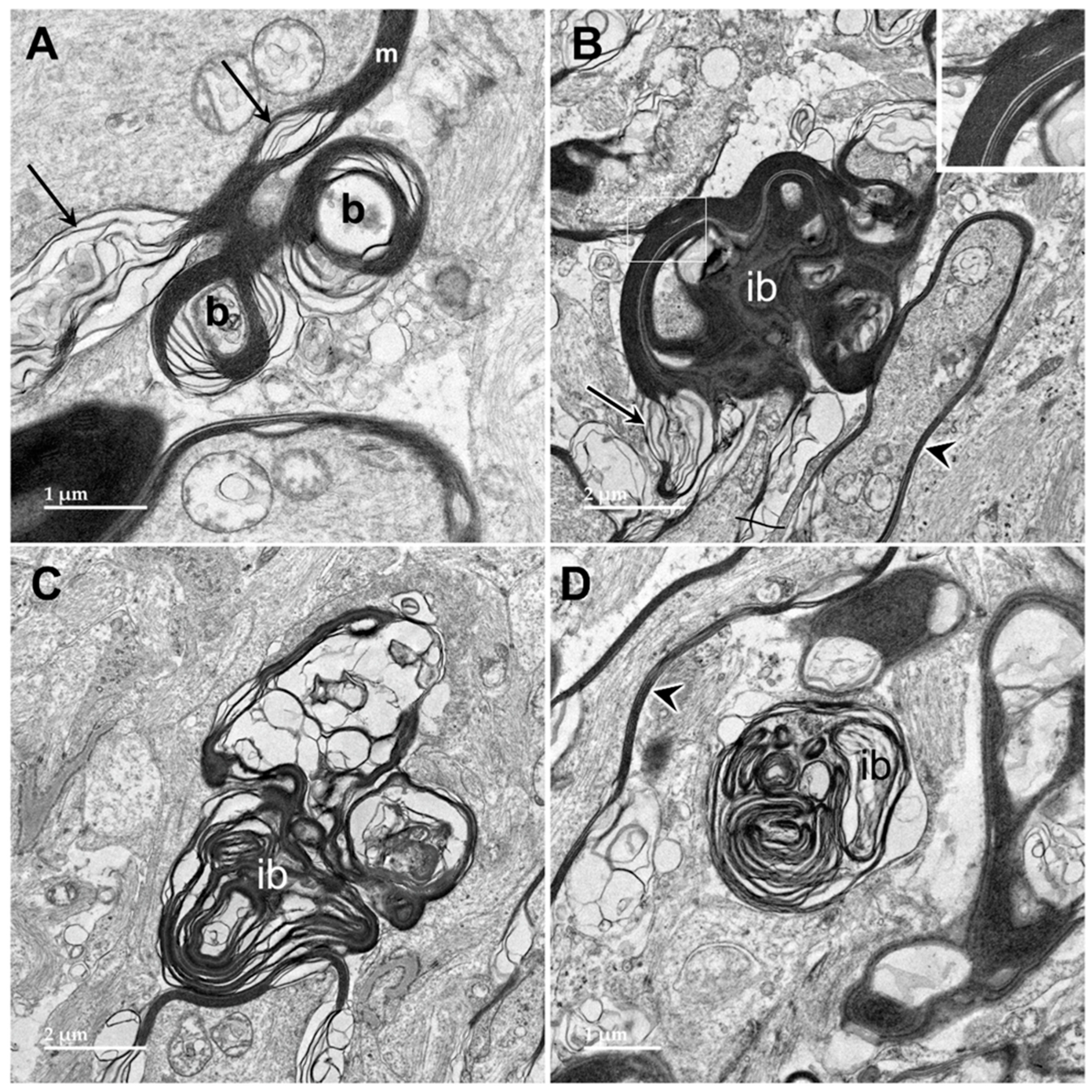
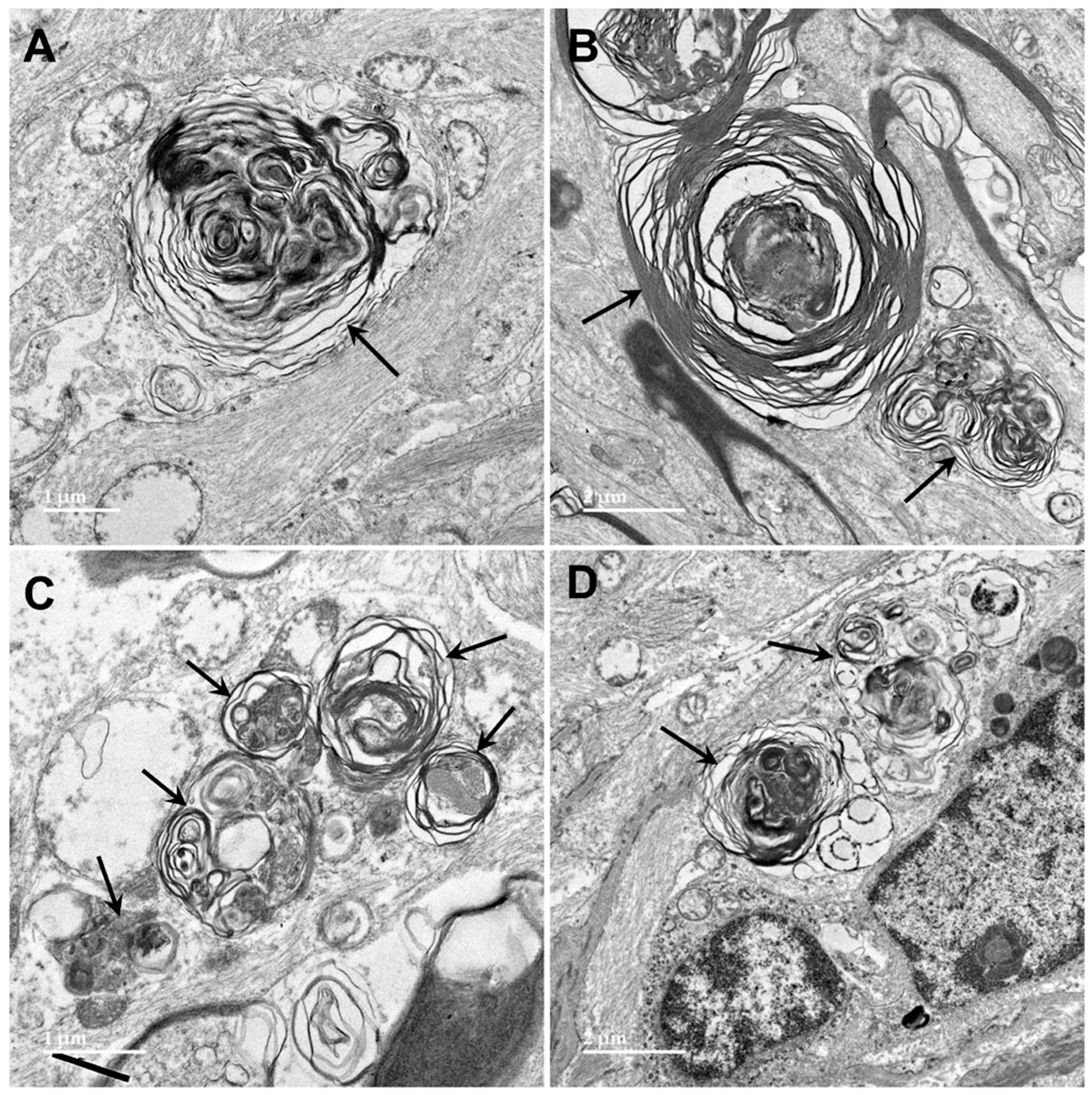
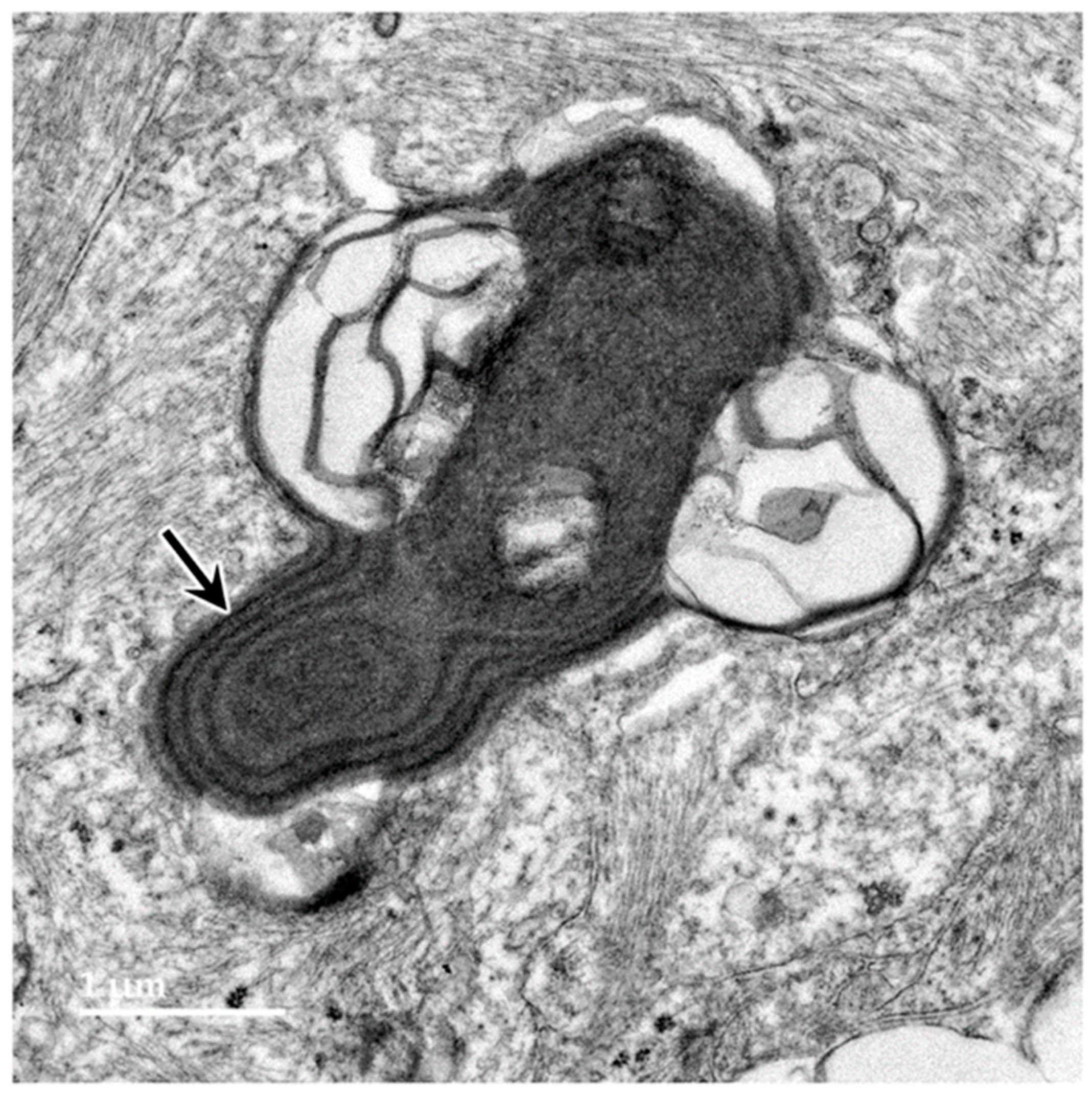
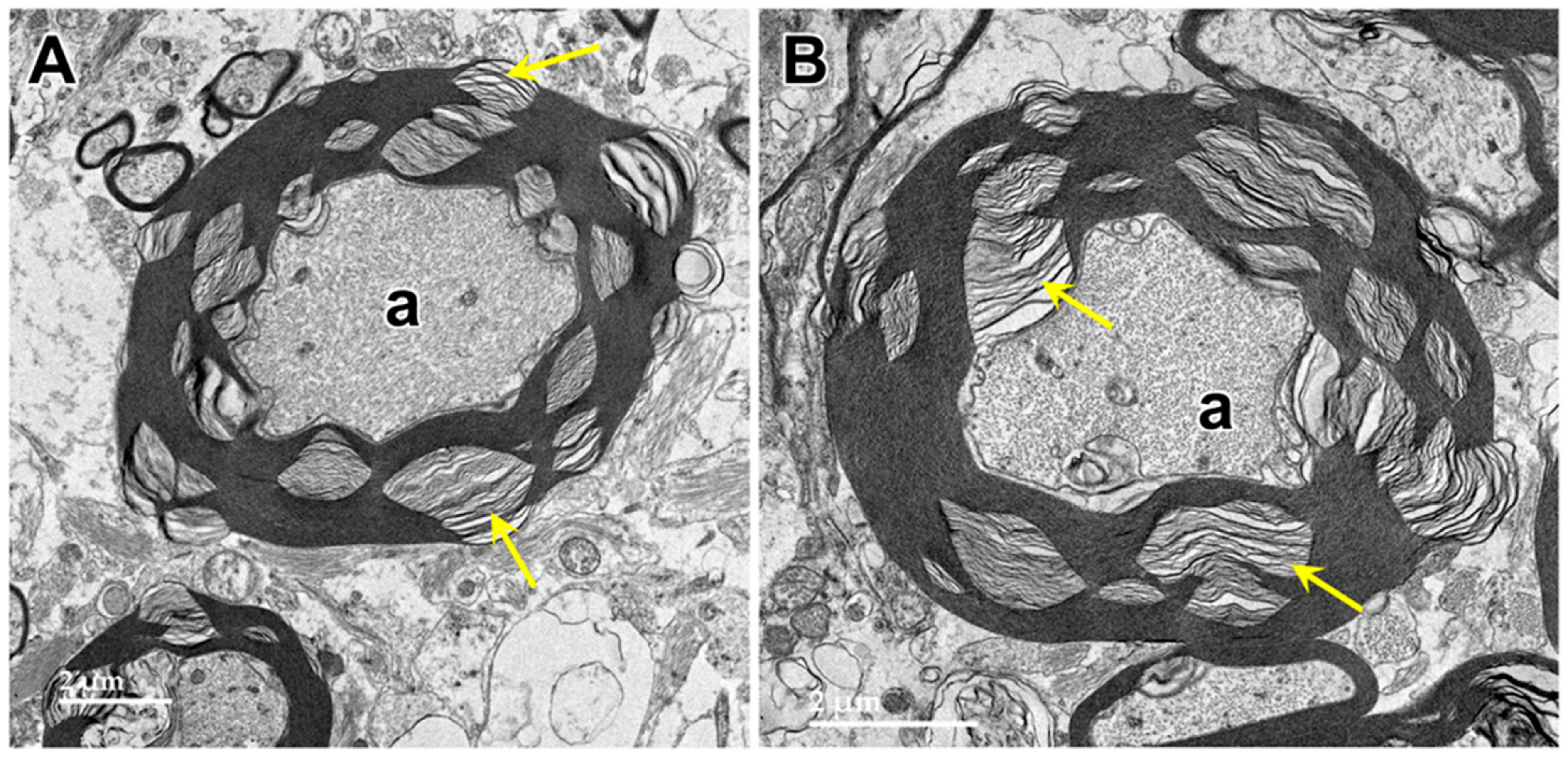
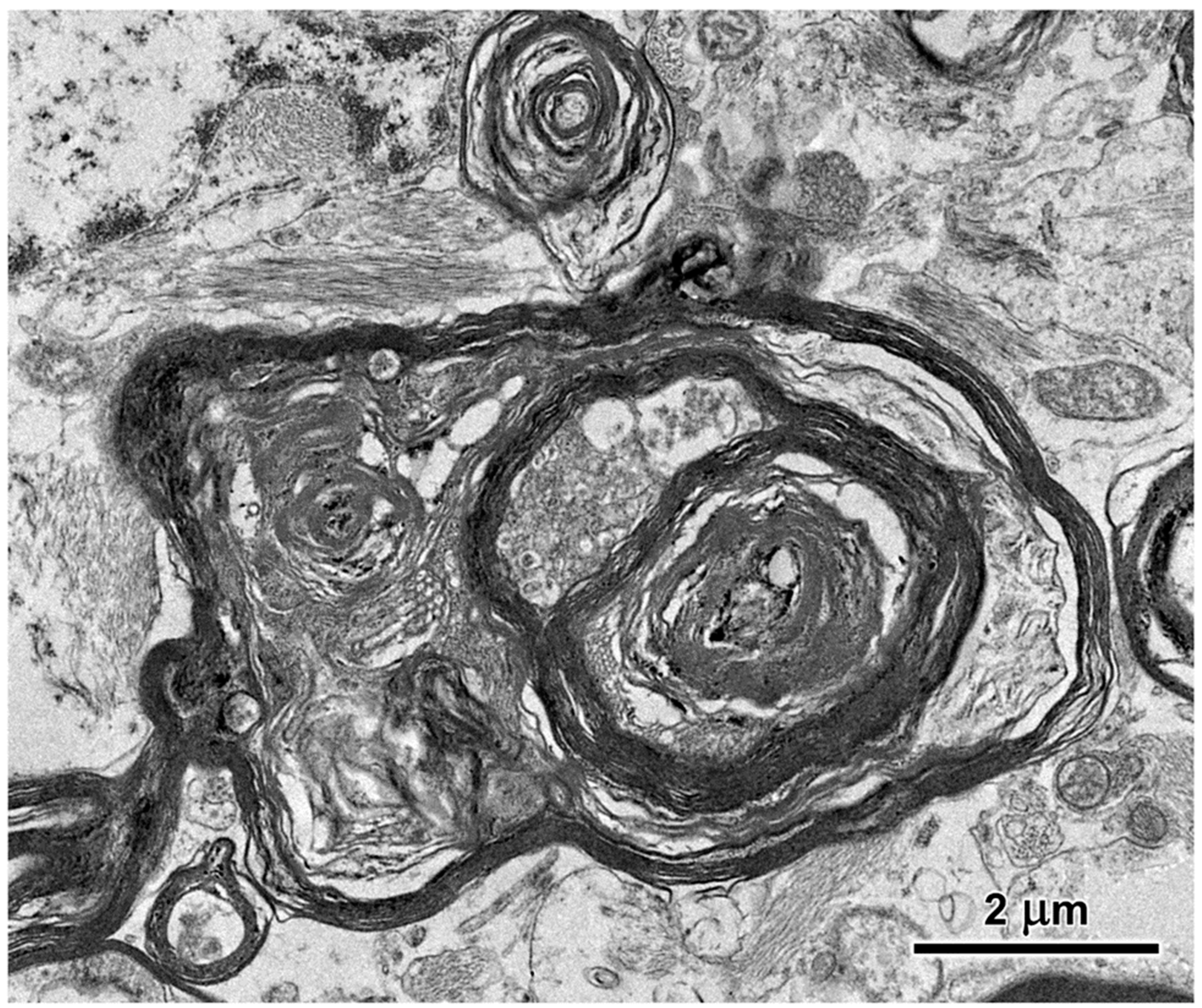
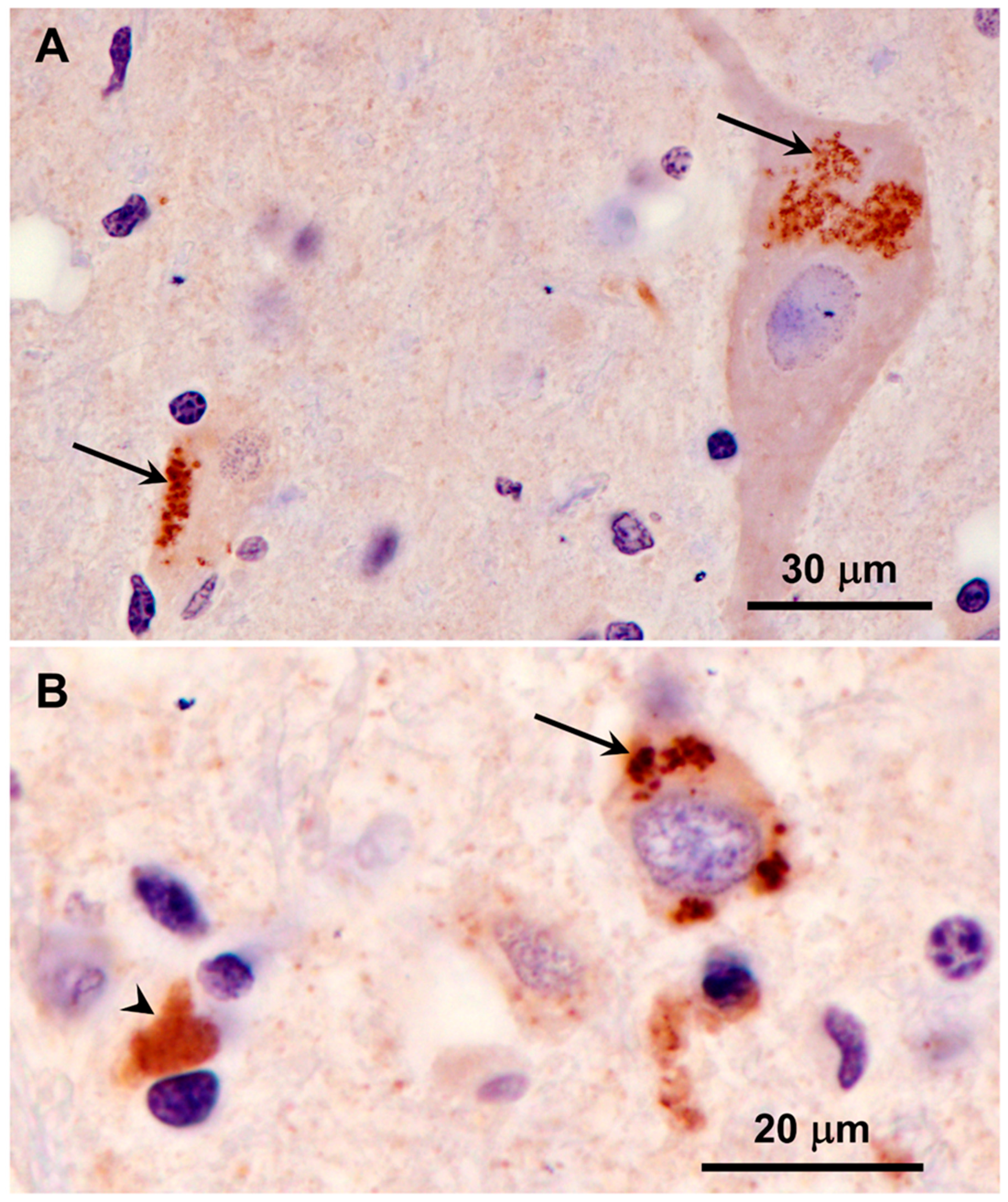
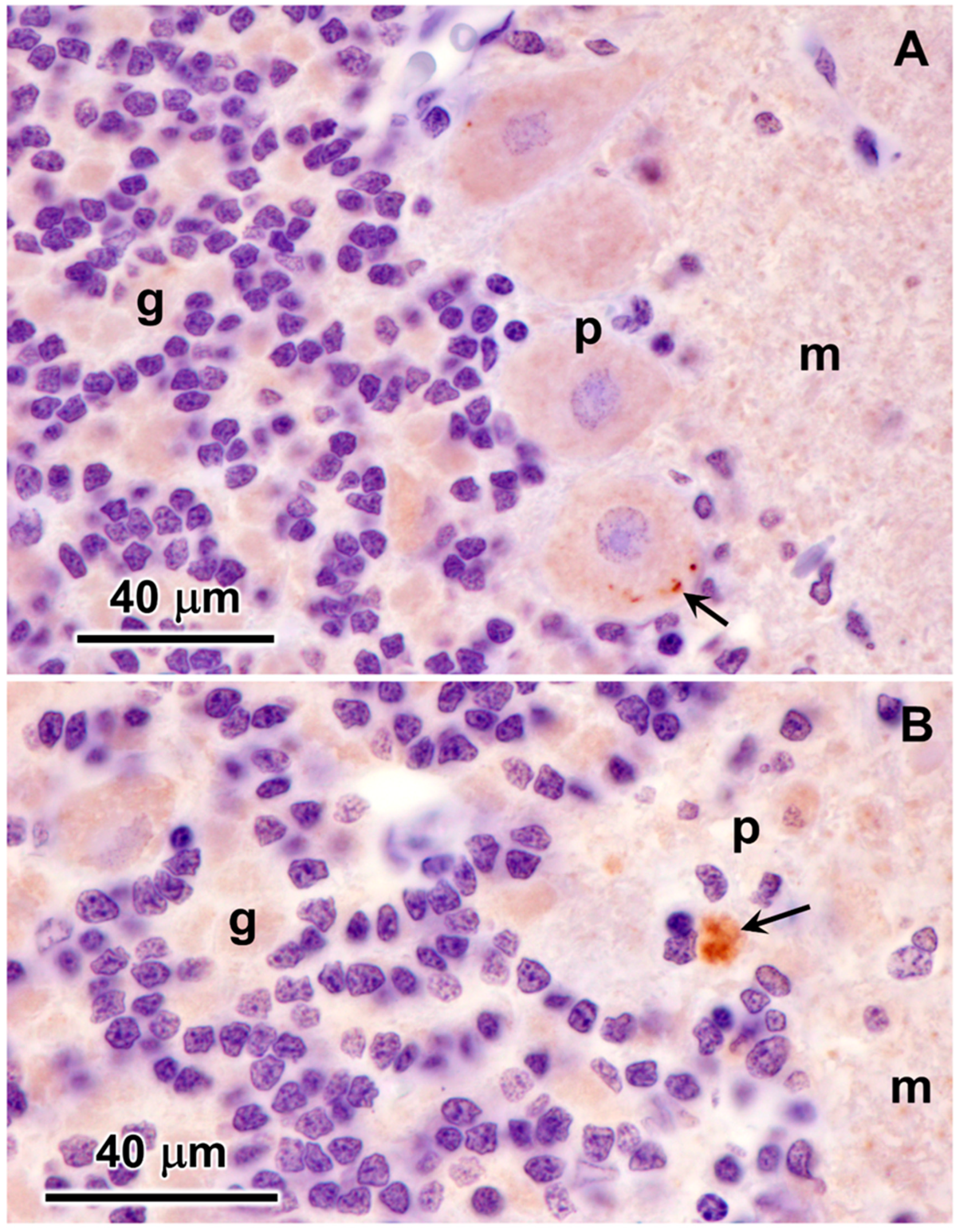
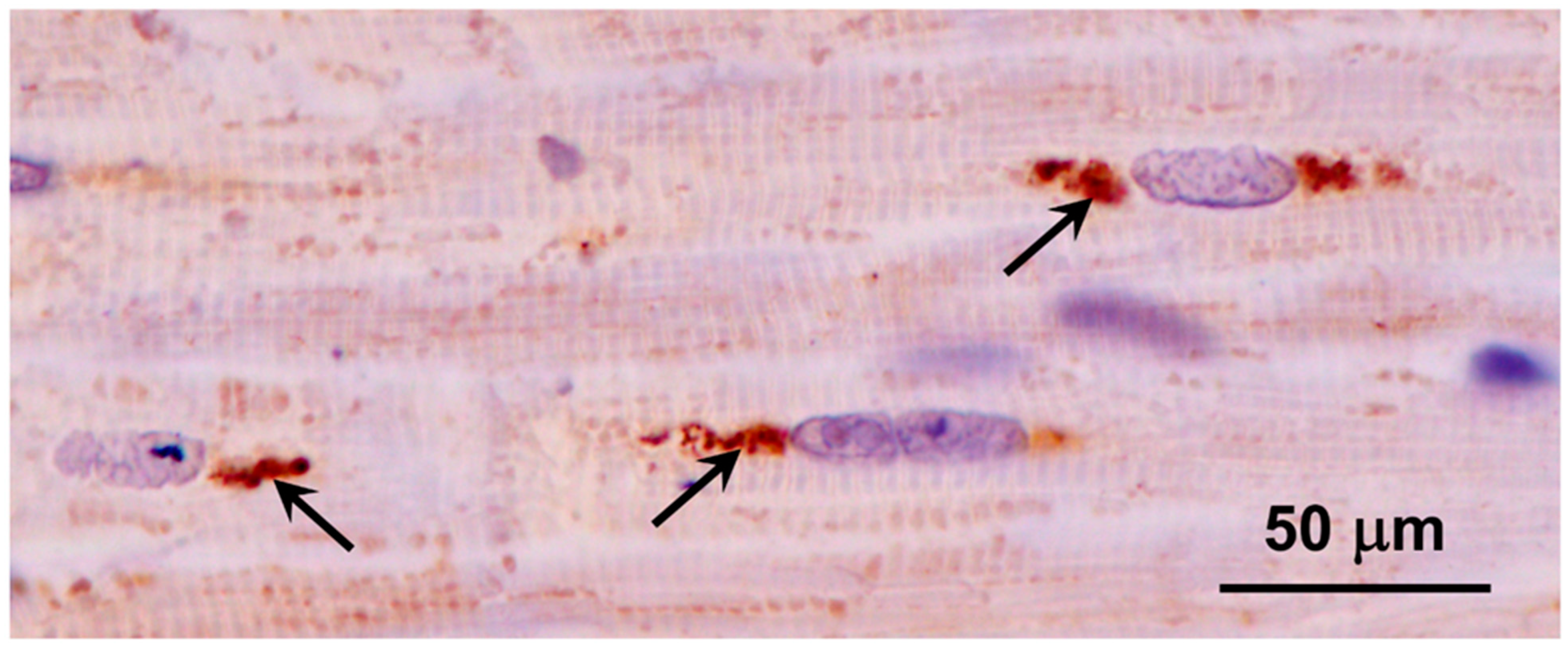
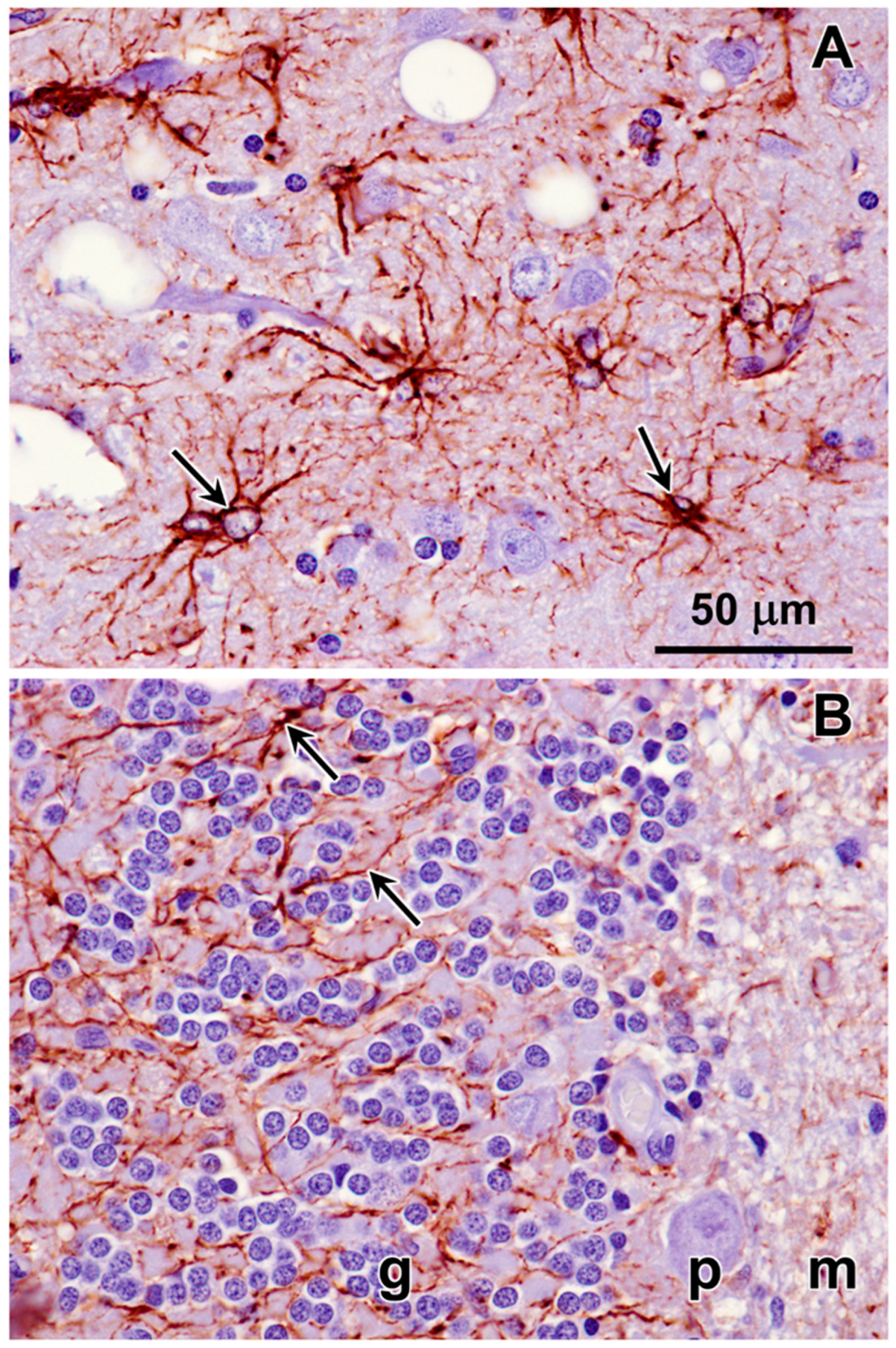
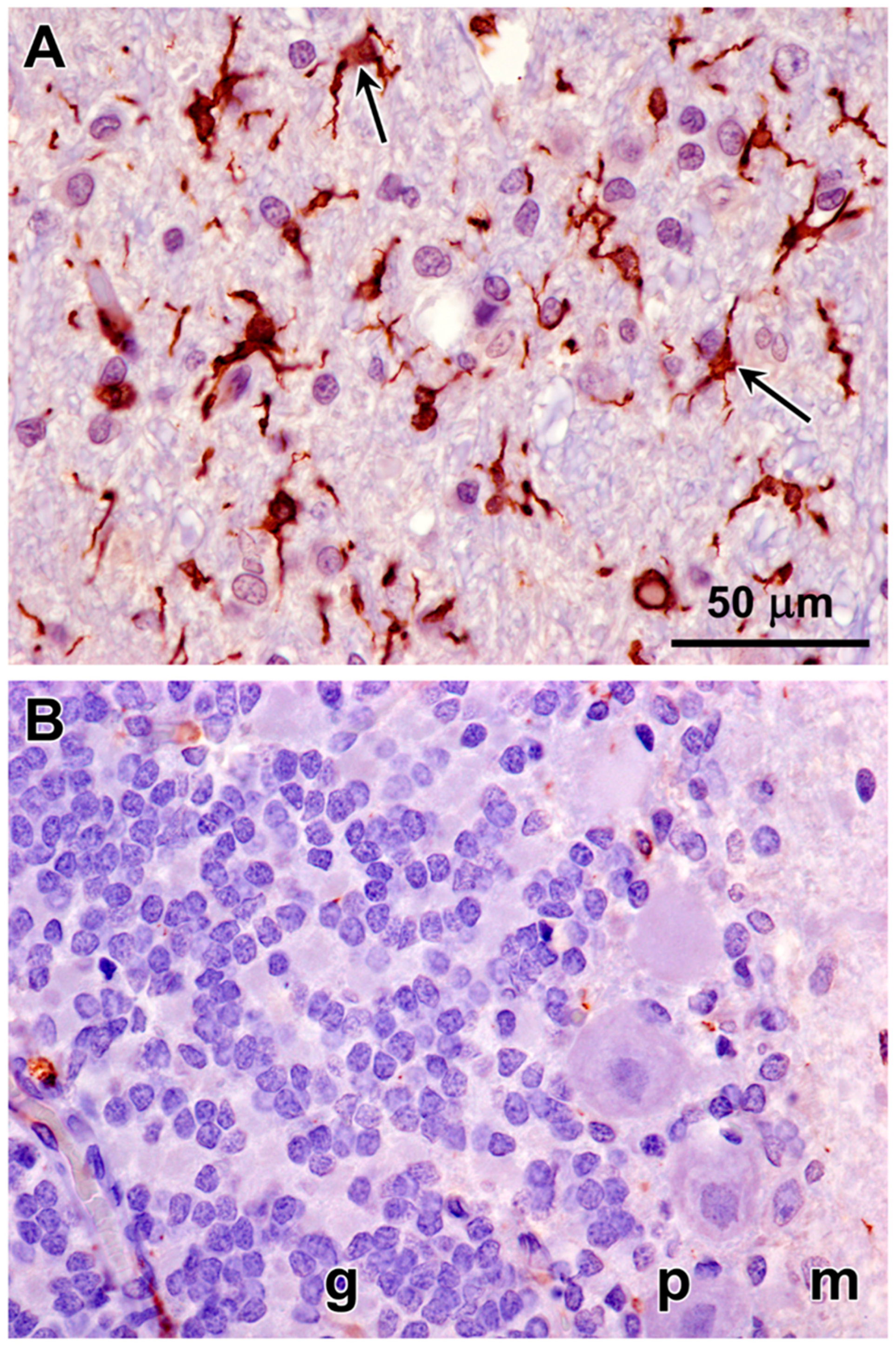
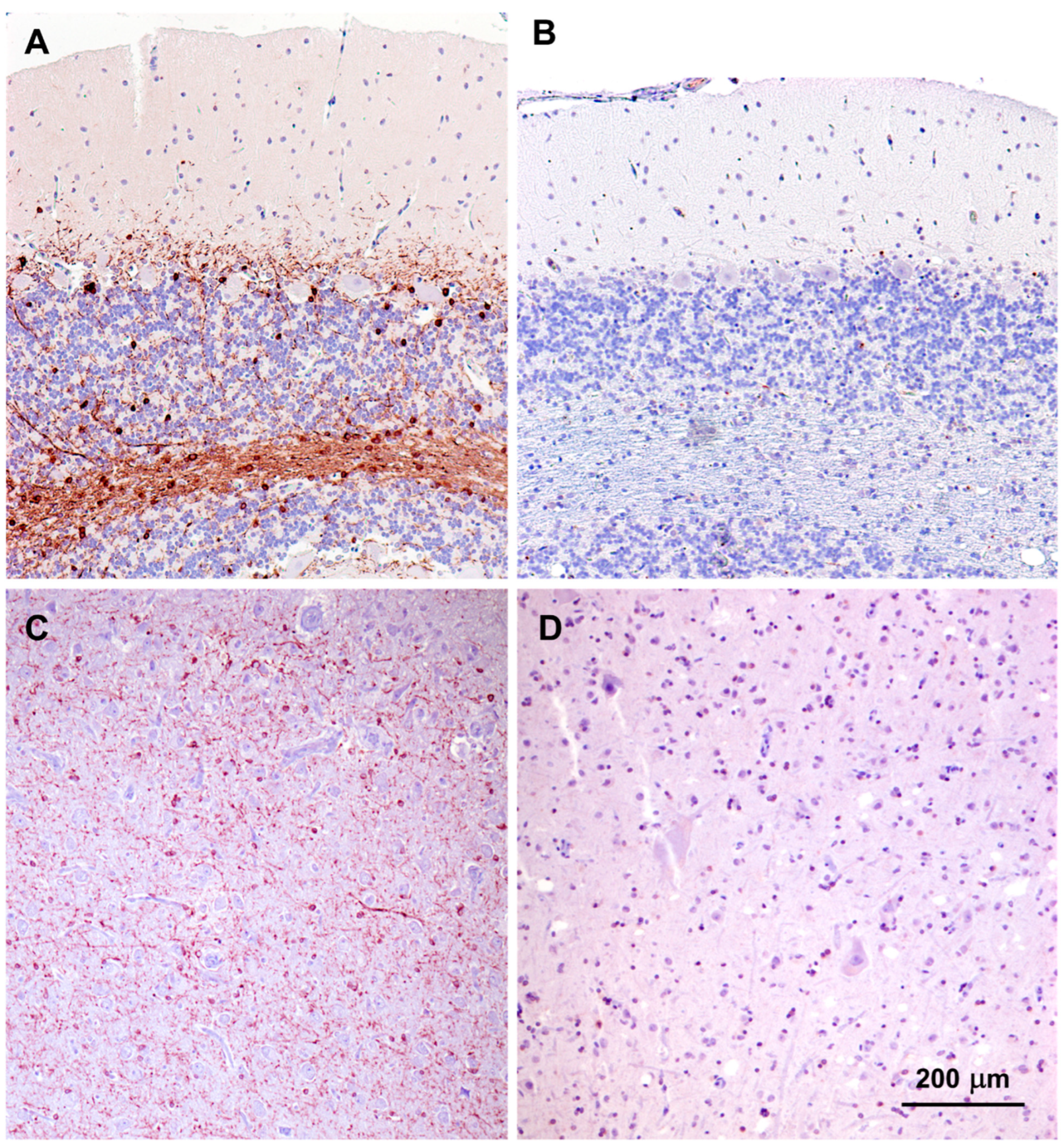
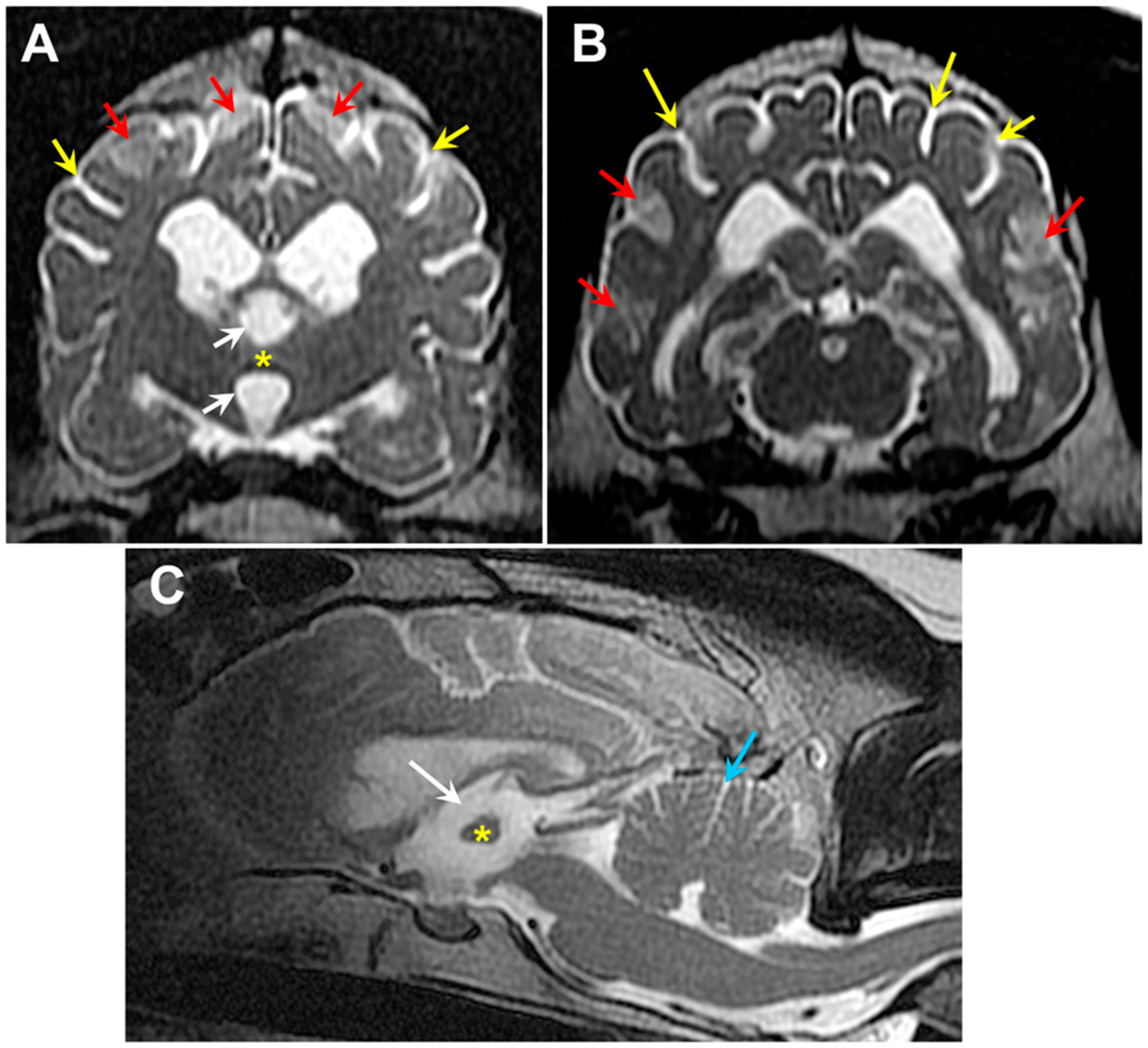
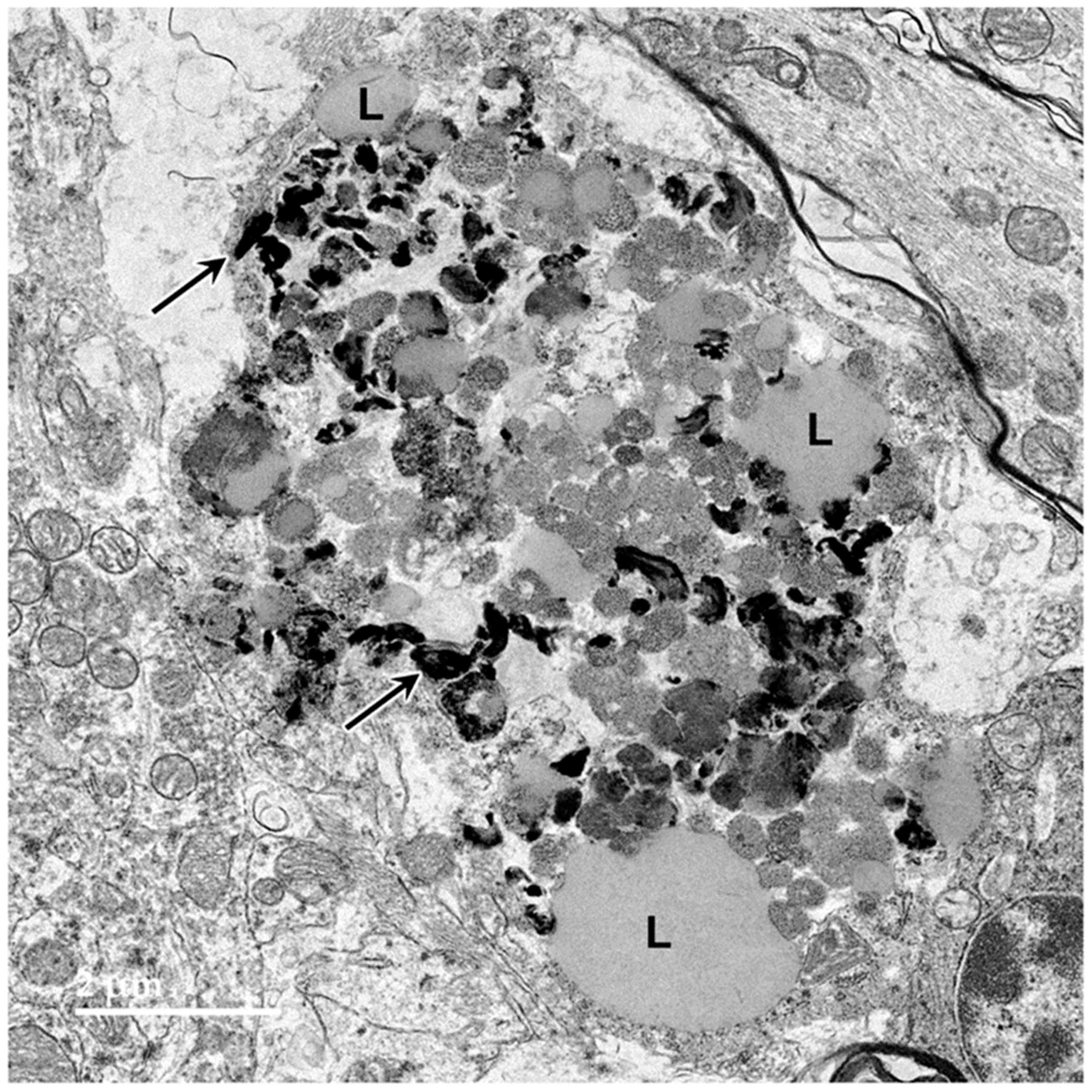
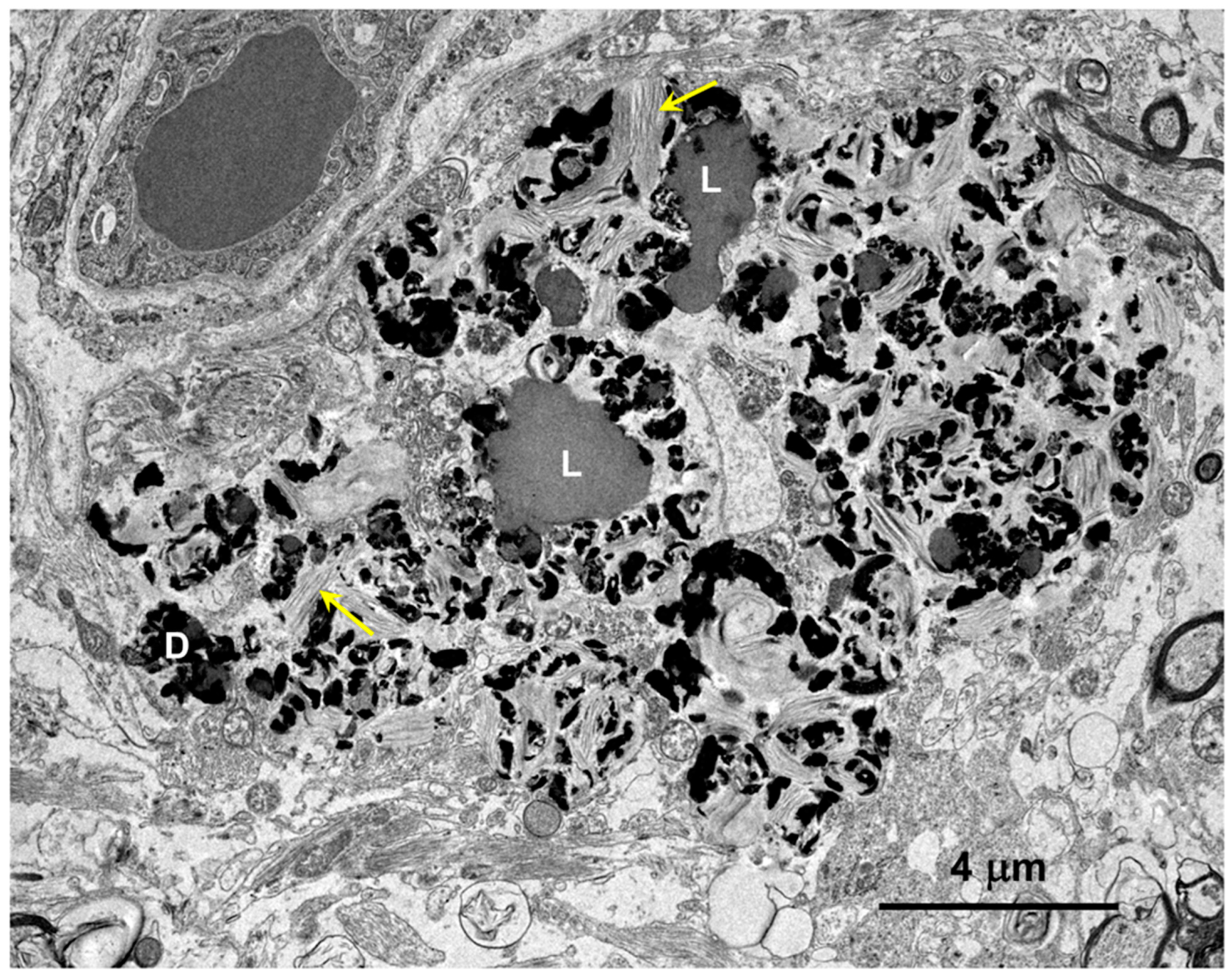
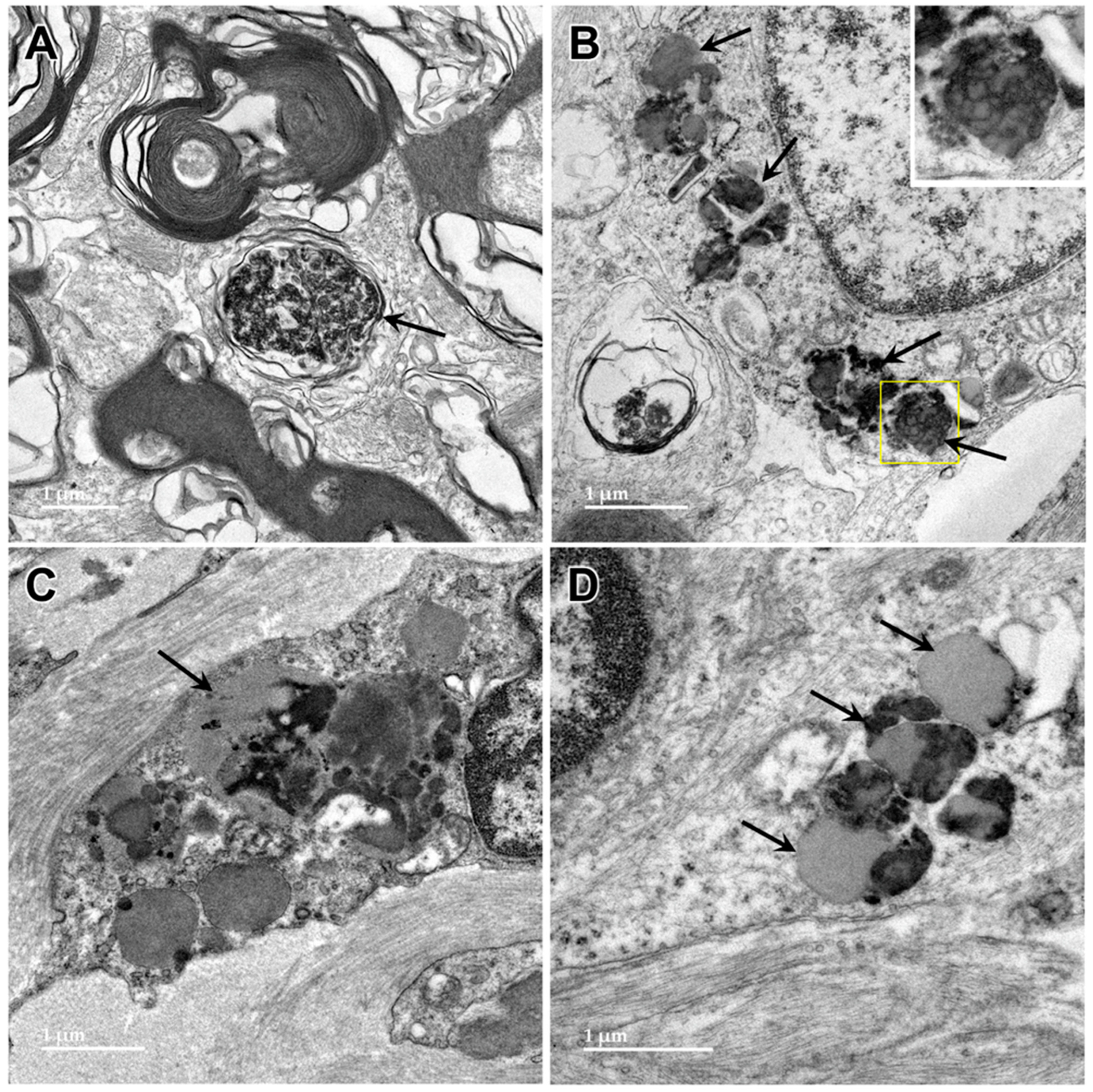
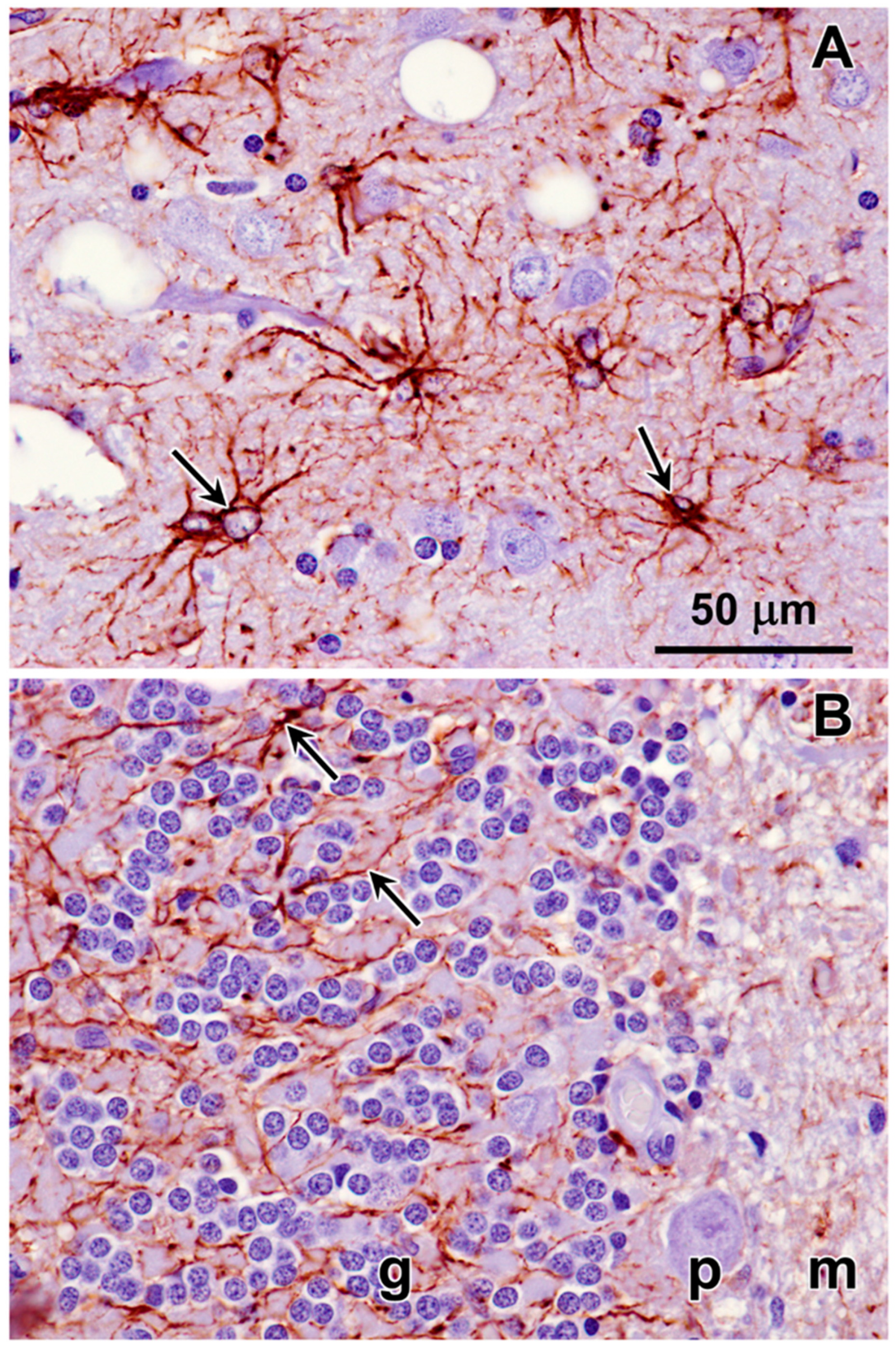
3.1. Microscopic Findings
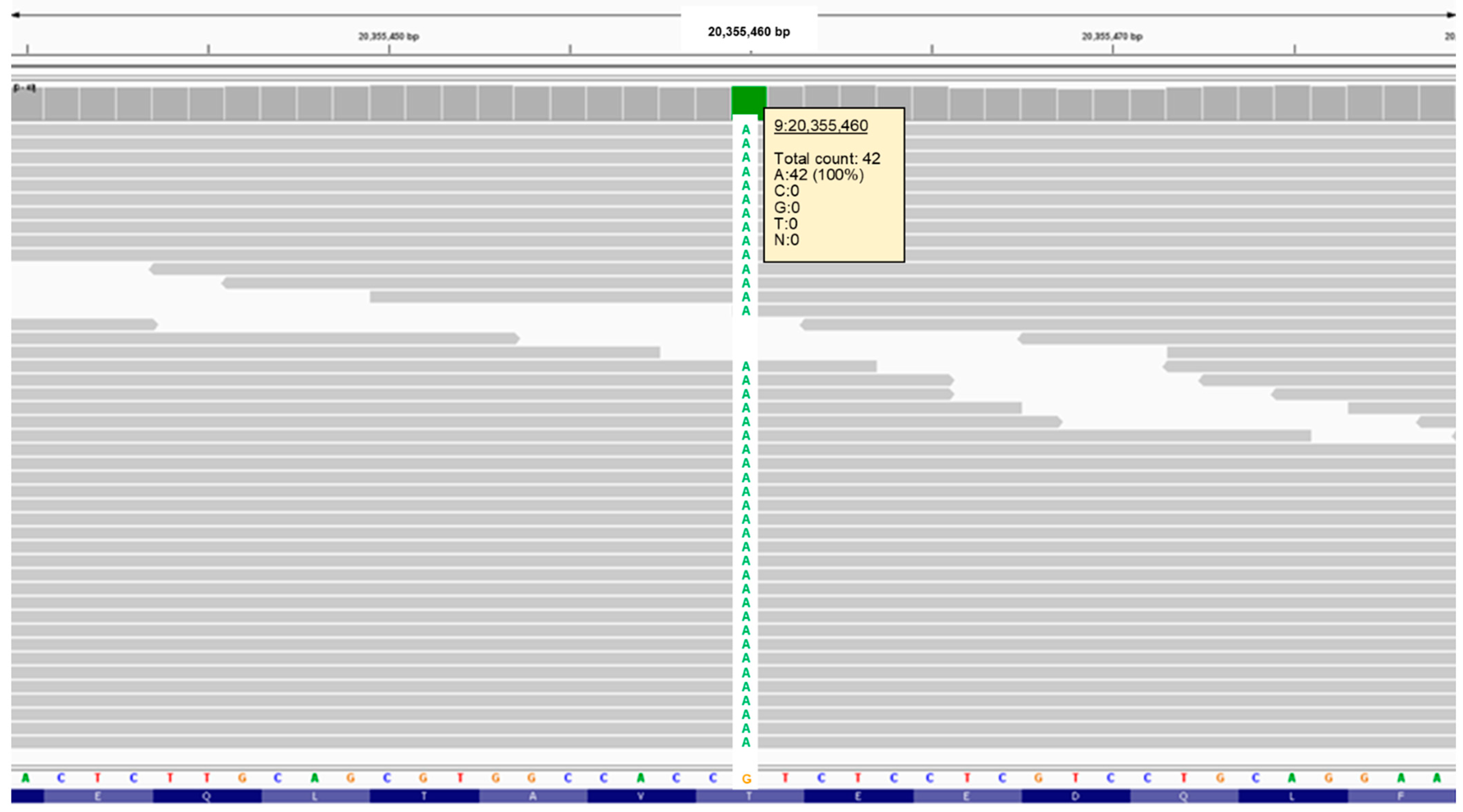
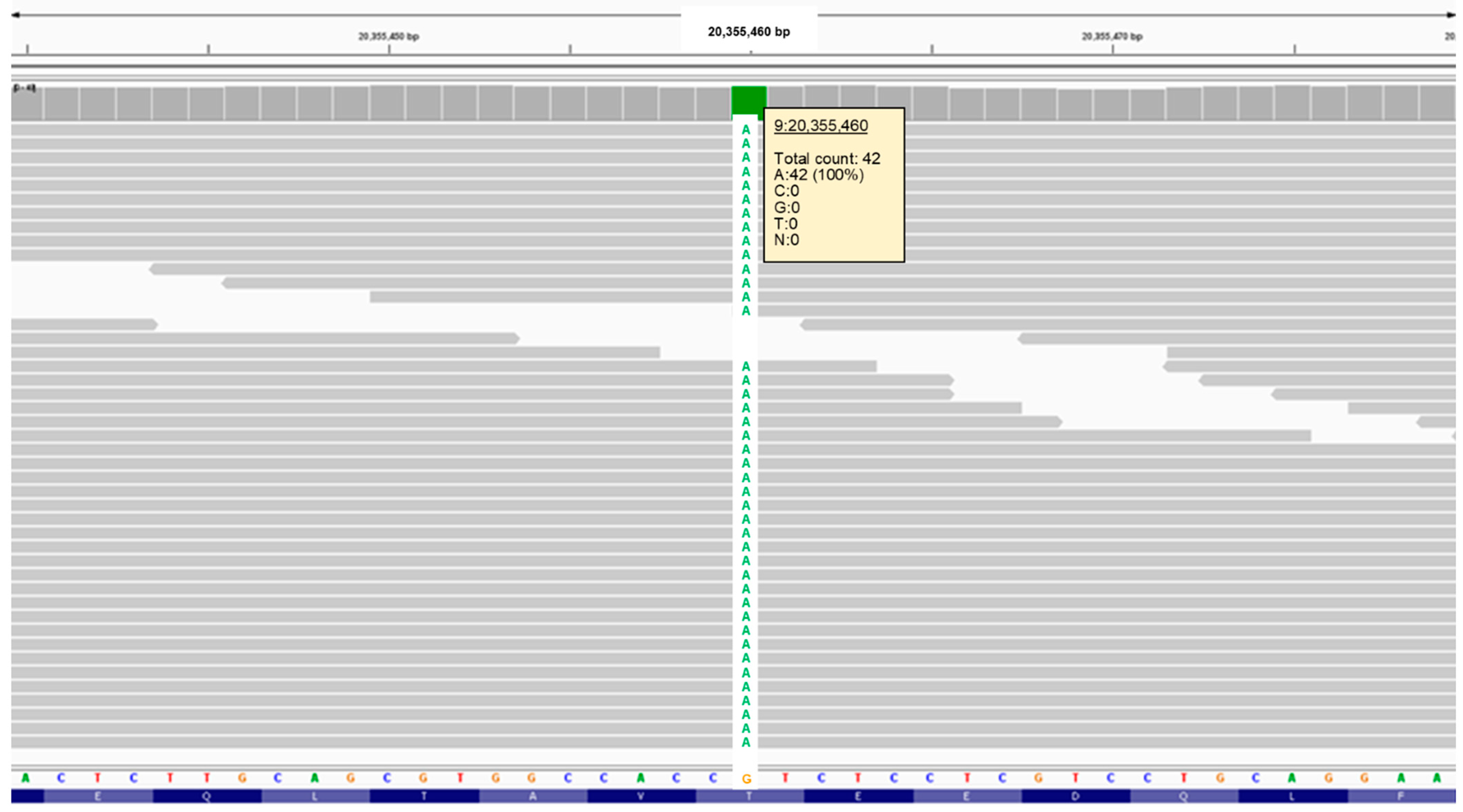
3.2. Molecular Genetic Findings
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bullock, G.; Johnson, G.S.; Mhlanga-Mutangadura, T.; Petesch, S.C.; Thompson, S.; Goebbels, S.; Katz, M.L. Lysosomal Storage Disease Associated with a CNP Sequence Variant in Dalmatian Dogs. Gene 2022, 830, 146513. [Google Scholar] [CrossRef]
- Bullock, G.; Johnson, G.S.; Pattridge, S.G.; Mhlanga-Mutangadura, T.; Guo, J.; Cook, J.; Campbell, R.S.; Vite, C.H.; Katz, M.L. A Homozygous MAN2B1 Missense Mutation in a Doberman Pinscher Dog with Neurodegeneration, Cytoplasmic Vacuoles, Autofluorescent Storage Granules, and an Alpha-Mannosidase Deficiency. Genes 2023, 14, 1746. [Google Scholar] [CrossRef]
- Morgan, B.R.; Coates, J.R.; Johnson, G.C.; Shelton, G.D.; Katz, M.L. Characterization of Thoracic Motor and Sensory Neurons and Spinal Nerve Roots in Canine Degenerative Myelopathy, a Potential Disease Model of Amyotrophic Lateral Sclerosis. J. Neurosci. Res. 2014, 92, 531–541. [Google Scholar] [CrossRef]
- Katz, M.L.; Khan, S.; Awano, T.; Shahid, S.A.; Siakotos, A.N.; Johnson, G.S. A Mutation in the CLN8 Gene in English Setter Dogs with Neuronal Ceroid-Lipofuscinosis. Biochem. Biophys. Res. Commun. 2005, 327, 541–547. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef]
- Jagannathan, V.; Hitte, C.; Kidd, J.M.; Masterson, P.; Murphy, T.D.; Emery, S.; Davis, B.; Buckley, R.M.; Liu, Y.-H.; Zhang, X.-Q.; et al. Dog10K_Boxer_Tasha_1.0: A Long-Read Assembly of the Dog Reference Genome. Genes 2021, 12, 847. [Google Scholar] [CrossRef]
- Hu, G.; Kurgan, L. Sequence Similarity Searching. Curr. Protoc. Protein Sci. 2019, 95, e71. [Google Scholar] [CrossRef]
- Coates, J.R.; Wininger, F.A. Canine Degenerative Myelopathy. Vet. Clin. N. Am. Small Anim. Pract. 2010, 40, 929–950. [Google Scholar] [CrossRef]
- Awano, T.; Johnson, G.S.; Wade, C.M.; Katz, M.L.; Johnson, G.C.; Taylor, J.F.; Perloski, M.; Biagi, T.; Baranowska, I.; Long, S.; et al. Genome-Wide Association Analysis Reveals a SOD1 Mutation in Canine Degenerative Myelopathy That Resemblesnamyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 2794–2799. [Google Scholar] [CrossRef]
- Zeng, R.; Coates, J.R.; Johnson, G.C.; Hansen, L.; Awano, T.; Kolicheski, A.; Ivansson, E.; Perloski, M.; Lindblad-Toh, K.; O’Brien, D.P.; et al. Breed Distribution of SOD1 Alleles Previously Associated with Canine Degenerative Myelopathy. J. Vet. Intern. Med. 2014, 28, 515–521. [Google Scholar] [CrossRef]
- Pemberton, T.J.; Choi, S.; Mayer, J.A.; Li, F.-Y.; Gokey, N.; Svaren, J.; Safra, N.; Bannasch, D.L.; Sullivan, K.; Breuhaus, B.; et al. A Mutation in the Canine Gene Encoding Folliculin-Interacting Protein 2 (FNIP2) Associated with a Unique Disruption in Spinal Cord Myelination. Glia 2014, 62, 39–51. [Google Scholar] [CrossRef]
- Safra, N.; Bassuk, A.G.; Ferguson, P.J.; Aguilar, M.; Coulson, R.L.; Thomas, N.; Hitchens, P.L.; Dickinson, P.J.; Vernau, K.M.; Wolf, Z.T.; et al. Genome-Wide Association Mapping in Dogs Enables Identification of the Homeobox Gene, NKX2-8, as a Genetic Component of Neural Tube Defects in Humans. PLoS Genet. 2013, 9, e1003646. [Google Scholar] [CrossRef]
- Katz, M.L.; Rustad, E.; Robinson, G.O.; Whiting, R.E.H.; Student, J.T.; Coates, J.R.; Narfstrom, K. Canine Neuronal Ceroid Lipofuscinoses: Promising Models for Preclinical Testing of Therapeutic Interventions. Neurobiol. Dis. 2017, 108, 277–287. [Google Scholar] [CrossRef]
- Anderson, G.W.; Goebel, H.H.; Simonati, A. Human Pathology in NCL. Biochim. Biophys. Acta 2013, 1832, 1807–1826. [Google Scholar] [CrossRef]
- Goebel, H.H. Morphological Aspects of the Neuronal Ceroid Lipofuscinoses. Neurol. Sci. 2000, 21, S27–S33. [Google Scholar] [CrossRef]
- Gotzl, J.K.; Mori, K.; Damme, M.; Fellerer, K.; Tahirovic, S.; Kleinberger, G.; Janssens, J.; van der Zee, J.; Lang, C.M.; Kremmer, E.; et al. Common Pathobiochemical Hallmarks of Progranulin-Associated Frontotemporal Lobar Degeneration and Neuronal Ceroid Lipofuscinosis. Acta Neuropathol. 2014, 127, 845–860. [Google Scholar] [CrossRef]
- Elleder, M.; Dvorakova, L.; Stolnaja, L.; Vlaskova, H.; Hulkova, H.; Druga, R.; Poupetova, H.; Kostalova, E.; Mikulastik, J. Atypical CLN2 with Later Onset and Prolonged Course: A Neuropathologic Study Showing Different Sensitivity of Neuronal Subpopulations to TPP1 Deficiency. Acta Neuropathol. 2008, 116, 119–124. [Google Scholar] [CrossRef]
- Goebel, H.H.; Kominami, E.; Neuen-Jacob, E.; Wheeler, R.B. Morphological Studies on CLN2. Eur. J. Paediatr. Neurol. 2001, 5 (Suppl. SA), 203–207. [Google Scholar] [CrossRef]
- Kurata, K.; Hayashi, M.; Satoh, J.; Kojima, H.; Nagata, J.; Tamagawa, K.; Shinohara, T.; Morimatsu, Y.; Kominami, E. Pathological Study on Sibling Autopsy Cases of the Late Infantile Form of Neuronal Ceroid Lipofuscinosis. Brain Dev. 1999, 21, 63–67. [Google Scholar] [CrossRef]
- Umehara, F.; Higuchi, I.; Tanaka, K.; Niiyama, T.; Ezaki, J.; Kominami, E.; Osame, M. Accumulation of Mitochondrial ATP Synthase Subunit c in Muscle in a Patient with Neuronal Ceroid Lipofuscinosis (Late Infantile Form). Acta Neuropathol. 1997, 93, 628–632. [Google Scholar] [CrossRef]
- Al-Abdi, L.; Al Murshedi, F.; Elmanzalawy, A.; Al Habsi, A.; Helaby, R.; Ganesh, A.; Ibrahim, N.; Patel, N.; Alkuraya, F.S. CNP Deficiency Causes Severe Hypomyelinating Leukodystrophy in Humans. Hum. Genet. 2020, 139, 615–622. [Google Scholar] [CrossRef]
- Lappe-Siefke, C.; Goebbels, S.; Gravel, M.; Nicksch, E.; Lee, J.; Braun, P.E.; Griffiths, I.R.; Nave, K.-A. Disruption of Cnp1 Uncouples Oligodendroglial Functions in Axonal Support and Myelination. Nat. Genet. 2003, 33, 366–374. [Google Scholar] [CrossRef]
- Rasband, M.N.; Tayler, J.; Kaga, Y.; Yang, Y.; Lappe-Siefke, C.; Nave, K.-A.; Bansal, R. CNP Is Required for Maintenance of Axon-Glia Interactions at Nodes of Ranvier in the CNS. Glia 2005, 50, 86–90. [Google Scholar] [CrossRef]
- Edgar, J.M.; McLaughlin, M.; Werner, H.B.; McCulloch, M.C.; Barrie, J.A.; Brown, A.; Faichney, A.B.; Snaidero, N.; Nave, K.-A.; Griffiths, I.R. Early Ultrastructural Defects of Axons and Axon-Glia Junctions in Mice Lacking Expression of Cnp1. Glia 2009, 57, 1815–1824. [Google Scholar] [CrossRef]
- Hagemeyer, N.; Goebbels, S.; Papiol, S.; Kastner, A.; Hofer, S.; Begemann, M.; Gerwig, U.C.; Boretius, S.; Wieser, G.L.; Ronnenberg, A.; et al. A Myelin Gene Causative of a Catatonia-Depression Syndrome upon Aging. EMBO Mol. Med. 2012, 4, 528–539. [Google Scholar] [CrossRef]
- Bouche, T.V.; Coates, J.R.; Moore, S.A.; Faissler, D.; Rishniw, M.; Olby, N.J. Diagnosis and Management of Dogs with Degenerative Myelopathy: A Survey of Neurologists and Rehabilitation Professionals. J. Vet. Intern. Med. 2023, 37, 1815–1820. [Google Scholar] [CrossRef]
- Kountourantzis, A.; Minoudi, S.; Karaiskou, N.; Papakostas, S.; Moulistanos, A.; Baka, R.D.; Tsartsianidou, V.; Vlachavas, A.; Aivaliotis, M.; Polizopoulou, Z.S.; et al. Prevalence of SOD1 Allele Associated with Degenerative Myelopathy in Canine Population in Greece. Res. Vet. Sci. 2023, 162, 104959. [Google Scholar] [CrossRef]
- Myllykoski, M.; Seidel, L.; Muruganandam, G.; Raasakka, A.; Torda, A.E.; Kursula, P. Structural and Functional Evolution of 2′,3′-Cyclic Nucleotide 3′-Phosphodiesterase. Brain Res. 2016, 1641, 64–78. [Google Scholar] [CrossRef]
- Olga, K.; Yulia, B.; Vassilios, P. The Functions of Mitochondrial 2′,3′-Cyclic Nucleotide-3′-Phosphodiesterase and Prospects for Its Future. Int. J. Mol. Sci. 2020, 21, 3217. [Google Scholar] [CrossRef]
- Vogel, U.S.; Thompson, R.J. Molecular Structure, Localization, and Possible Functions of the Myelin-Associated Enzyme 2′,3′-Cyclic Nucleotide 3′-Phosphodiesterase. J. Neurochem. 1988, 50, 1667–1677. [Google Scholar] [CrossRef]
- Jackson, E.K. Discovery and Roles of 2′,3′-cAMP in Biological Systems. Handb. Exp. Pharmacol. 2017, 238, 229–252. [Google Scholar] [CrossRef]
- Siems, S.B.; Jahn, O.; Eichel, M.A.; Kannaiyan, N.; Wu, L.M.N.; Sherman, D.L.; Kusch, K.; Hesse, D.; Jung, R.B.; Fledrich, R.; et al. Proteome Profile of Peripheral Myelin in Healthy Mice and in a Neuropathy Model. Elife 2020, 9, e51406. [Google Scholar] [CrossRef]
- Kursula, P. The Current Status of Structural Studies on Proteins of the Myelin Sheath (Review). Int. J. Mol. Med. 2001, 8, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Raasakka, A.; Kursula, P. The Myelin Membrane-Associated Enzyme 2′,3′-Cyclic Nucleotide 3′-Phosphodiesterase: On a Highway to Structure and Function. Neurosci. Bull. 2014, 30, 956–966. [Google Scholar] [CrossRef]
- O’Neill, R.C.; Minuk, J.; Cox, M.E.; Braun, P.E.; Gravel, M. CNP2 mRNA Directs Synthesis of Both CNP1 and CNP2 Polypeptides. J. Neurosci. Res. 1997, 50, 248–257. [Google Scholar] [CrossRef]
- Kurihara, T.; Monoh, K.; Sakimura, K.; Takahashi, Y. Alternative Splicing of Mouse Brain 2′,3′-Cyclic-Nucleotide 3′-Phosphodiesterase mRNA. Biochem. Biophys. Res. Commun. 1990, 170, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; O’Neill, R.C.; Park, M.W.; Gravel, M.; Braun, P.E. Mitochondrial Localization of CNP2 Is Regulated by Phosphorylation of the N-terminal Targeting Signal by PKC: Implications of a Mitochondrial Function for CNP2 in Glial and Non-Glial Cells. Mol. Cell Neurosci. 2006, 31, 446–462. [Google Scholar] [CrossRef]
- Baburina, Y.; Odinokova, I.; Azarashvili, T.; Akatov, V.; Sotnikova, L.; Krestinina, O. Possible Involvement of 2′,3′-Cyclic Nucleotide-3′-Phosphodiesterase in the Protein Phosphorylation-Mediated Regulation of the Permeability Transition Pore. Int. J. Mol. Sci. 2018, 19, 3499. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Yu, Q.; Zhang, L.; Jiang, Z. Cyclophilin D-Mediated Mitochondrial Permeability Transition Regulates Mitochondrial Function. Curr. Pharm. Des. 2023, 29, 620–629. [Google Scholar] [CrossRef]
- Mazzeo, A.T.; Beat, A.; Singh, A.; Bullock, M.R. The Role of Mitochondrial Transition Pore, and Its Modulation, in Traumatic Brain Injury and Delayed Neurodegeneration after TBI. Exp. Neurol. 2009, 218, 363–370. [Google Scholar] [CrossRef]
- Robichaux, D.J.; Harata, M.; Murphy, E.; Karch, J. Mitochondrial Permeability Transition Pore-Dependent Necrosis. J. Mol. Cell Cardiol. 2023, 174, 47–55. [Google Scholar] [CrossRef]
- Bernardi, P.; Gerle, C.; Halestrap, A.P.; Jonas, E.A.; Karch, J.; Mnatsakanyan, N.; Pavlov, E.; Sheu, S.-S.; Soukas, A.A. Identity, Structure, and Function of the Mitochondrial Permeability Transition Pore: Controversies, Consensus, Recent Advances, and Future Directions. Cell Death Differ. 2023, 30, 1869–1885. [Google Scholar] [CrossRef]
- Mole, S.E.; Williams, R.E.; Goebel, H.H. The Neuronal Ceroid Lipofuscinoses (Batten Disease), 2nd ed.; Mole, S.E., Willimas, R.E., Goebel, H.H., Eds.; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Palmer, D.N.; Barry, L.A.; Tyynela, J.; Cooper, J.D. NCL Disease Mechanisms. Biochim. Biophys. Acta 2013, 1832, 1882–1893. [Google Scholar] [CrossRef]




















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keller, S.H.; Johnson, G.S.; Bullock, G.; Mhlanga-Mutangadura, T.; Schwartz, M.; Pattridge, S.G.; Guo, J.; Kortz, G.D.; Katz, M.L. Homozygous CNP Mutation and Neurodegeneration in Weimaraners: Myelin Abnormalities and Accumulation of Lipofuscin-like Inclusions. Genes 2024, 15, 246. https://doi.org/10.3390/genes15020246
Keller SH, Johnson GS, Bullock G, Mhlanga-Mutangadura T, Schwartz M, Pattridge SG, Guo J, Kortz GD, Katz ML. Homozygous CNP Mutation and Neurodegeneration in Weimaraners: Myelin Abnormalities and Accumulation of Lipofuscin-like Inclusions. Genes. 2024; 15(2):246. https://doi.org/10.3390/genes15020246
Chicago/Turabian StyleKeller, Stefan H., Gary S. Johnson, Garrett Bullock, Tendai Mhlanga-Mutangadura, Malte Schwartz, Savannah G. Pattridge, Juyuan Guo, Gregg D. Kortz, and Martin L. Katz. 2024. "Homozygous CNP Mutation and Neurodegeneration in Weimaraners: Myelin Abnormalities and Accumulation of Lipofuscin-like Inclusions" Genes 15, no. 2: 246. https://doi.org/10.3390/genes15020246
APA StyleKeller, S. H., Johnson, G. S., Bullock, G., Mhlanga-Mutangadura, T., Schwartz, M., Pattridge, S. G., Guo, J., Kortz, G. D., & Katz, M. L. (2024). Homozygous CNP Mutation and Neurodegeneration in Weimaraners: Myelin Abnormalities and Accumulation of Lipofuscin-like Inclusions. Genes, 15(2), 246. https://doi.org/10.3390/genes15020246