
Characteristics and Comparative Analysis of the Special-Structure (Non-Single-Circle) Mitochondrial Genome of Capsicum pubescens Ruiz & Pav

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. DNA Sequencing, Genome Assembly, and Annotation
2.3. Codon Usage, Selective Pressure, RNA Editing Prediction, and Repeat Sequence Identification
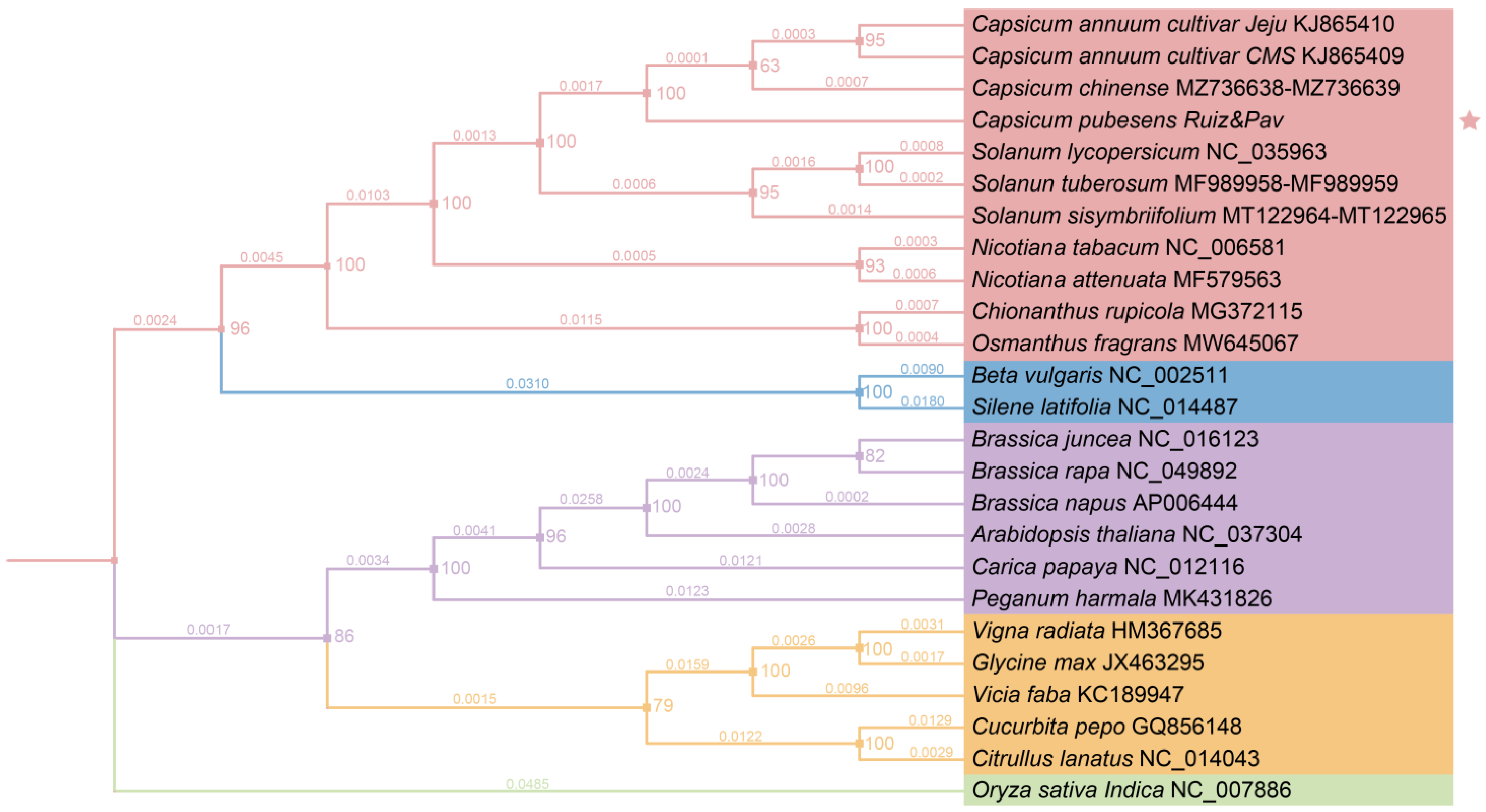
2.4. Phylogenetic Analysis
3. Results and Discussion
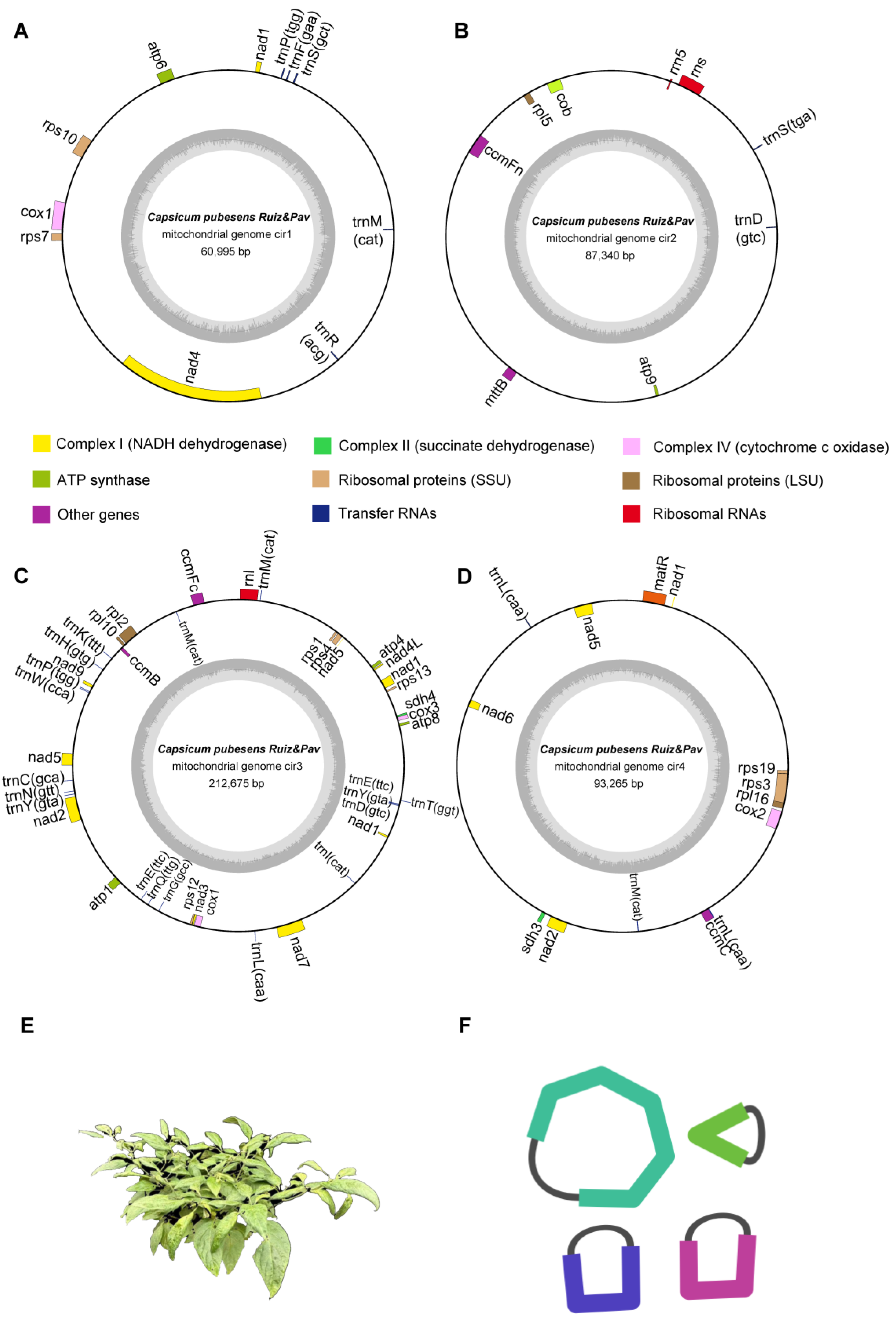
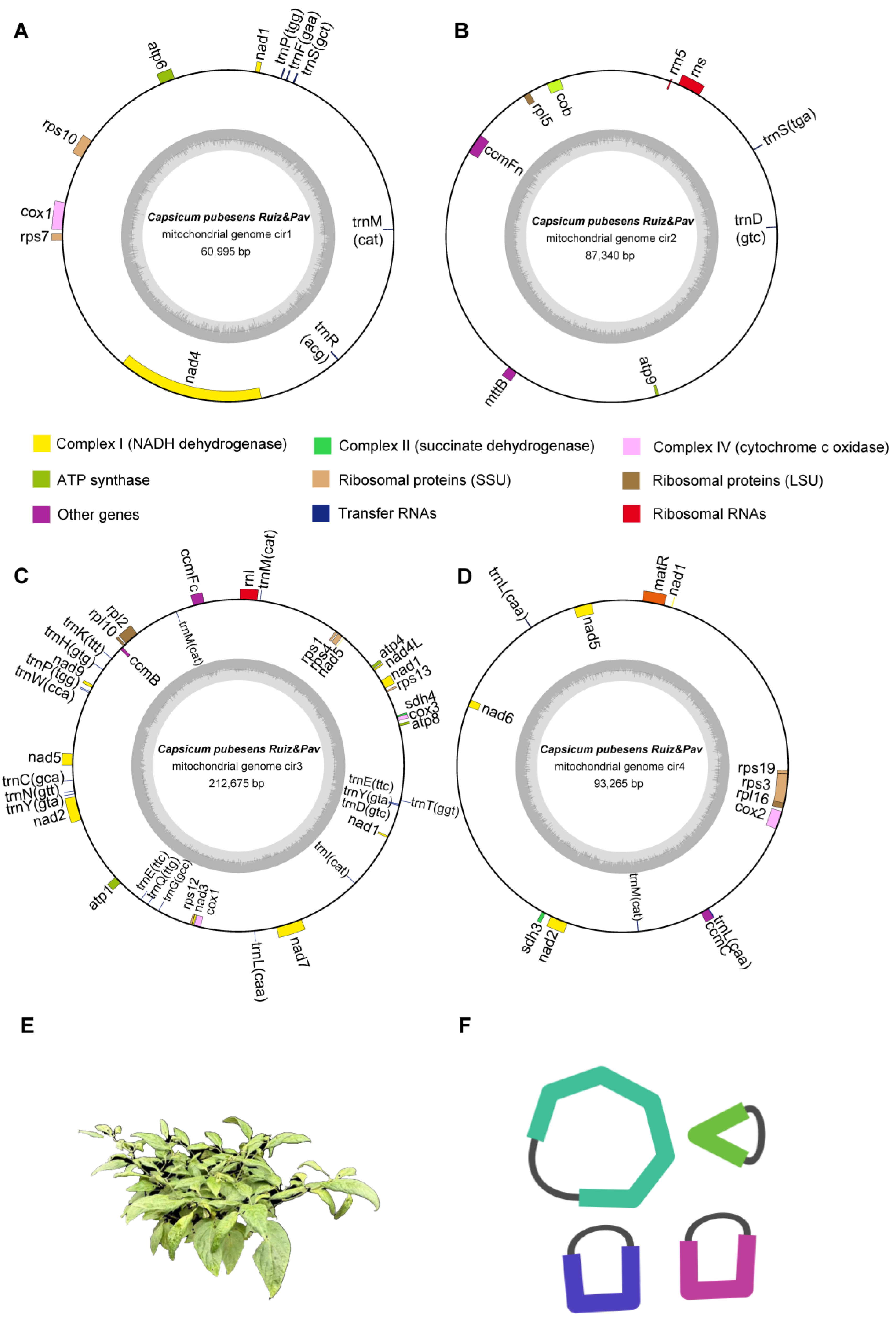
3.1. Genomic Features of the C. pubescens Mitochondrial Genome
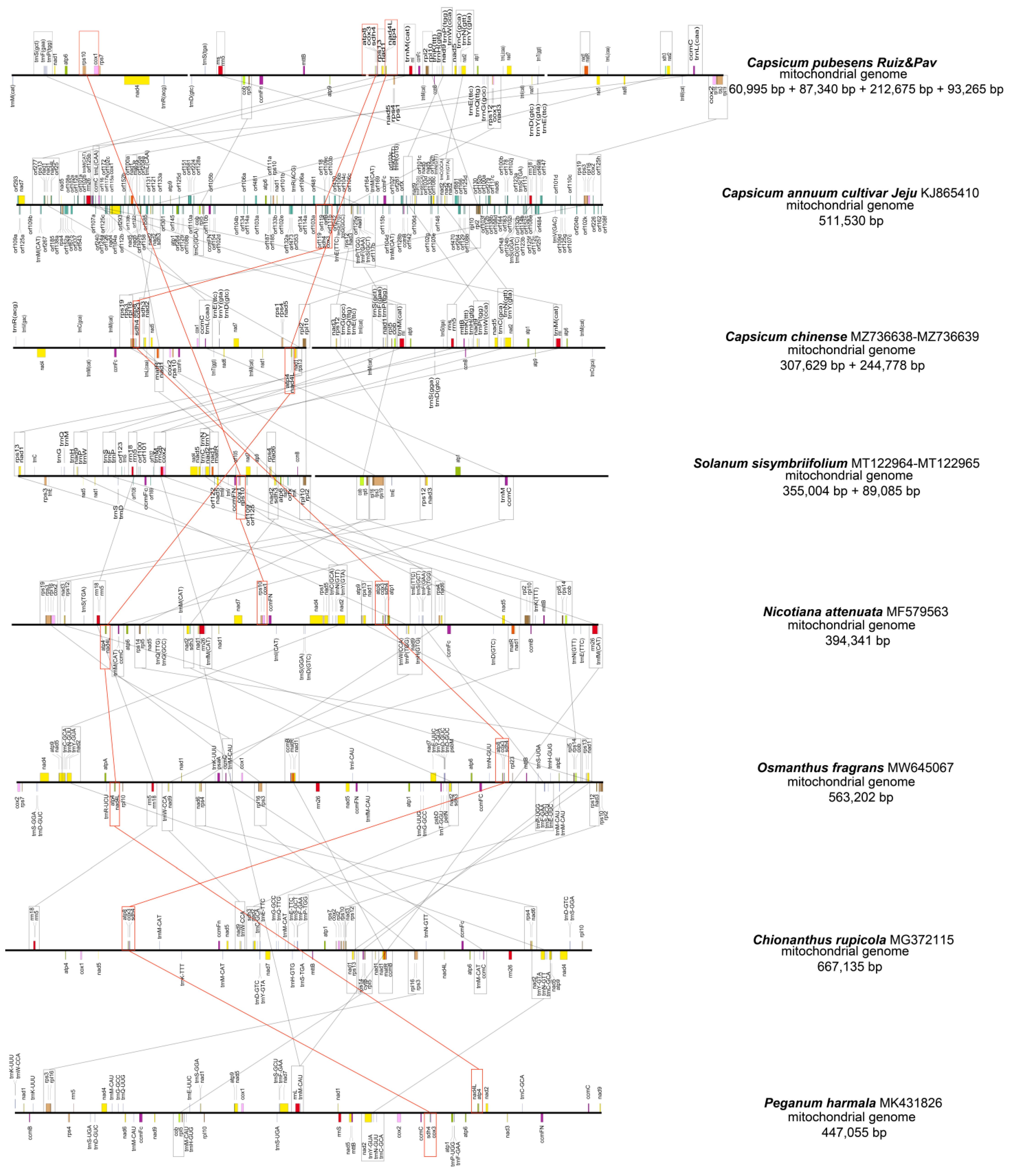
3.1.1. Mitochondrial Structure and Gene Content
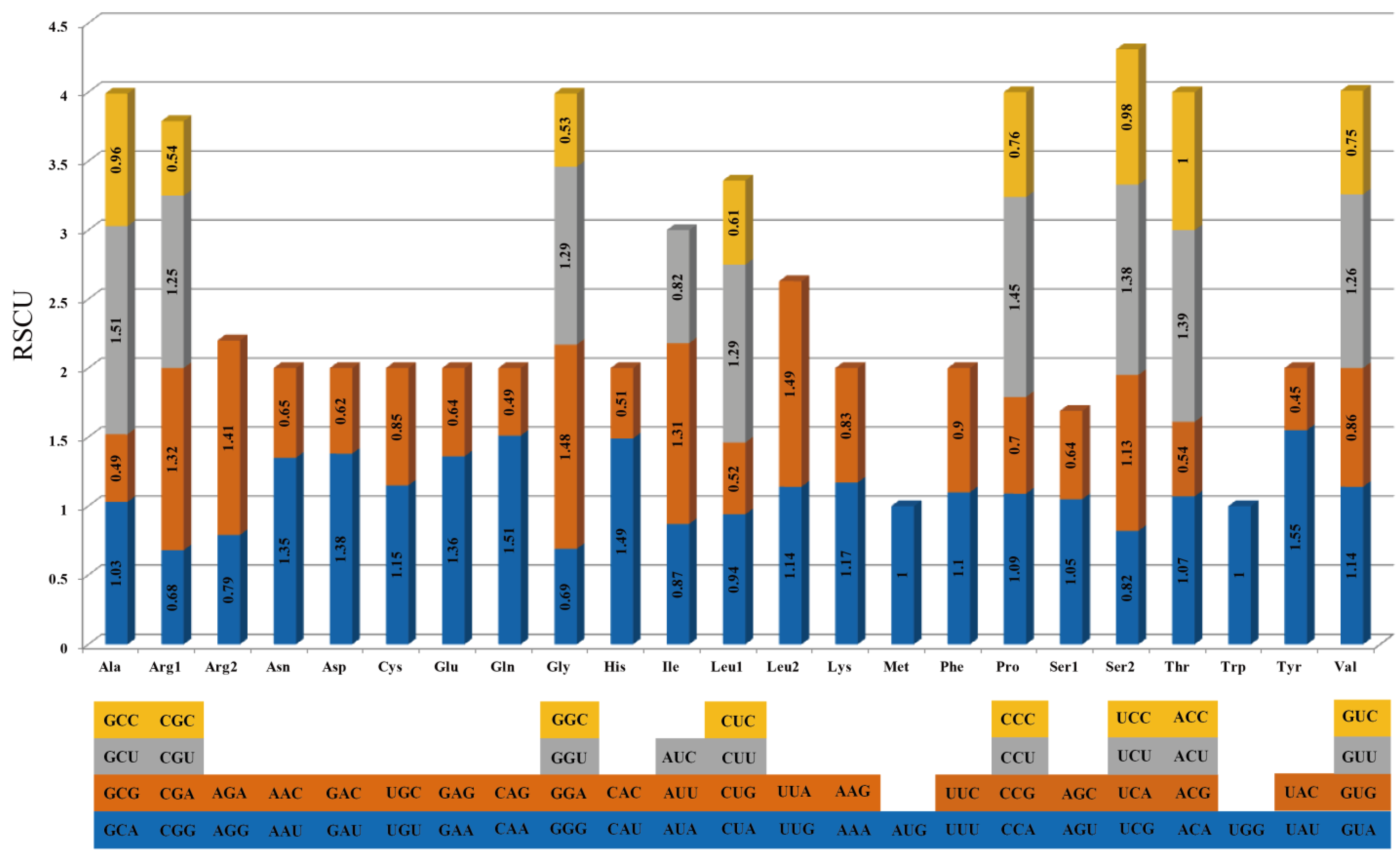
3.1.2. Codon Usage Analysis of PCGs
3.1.3. Repeat Sequences Analysis
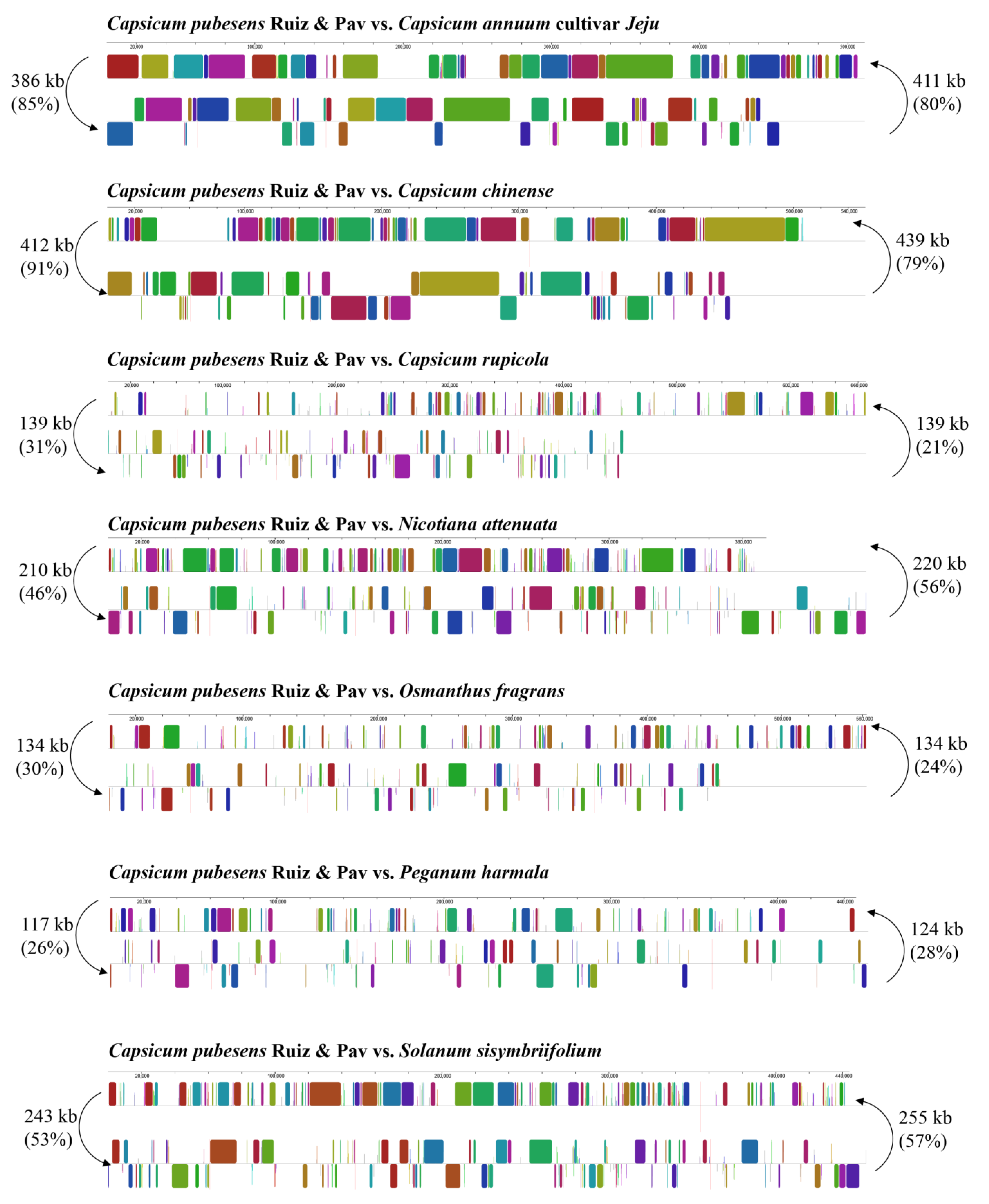
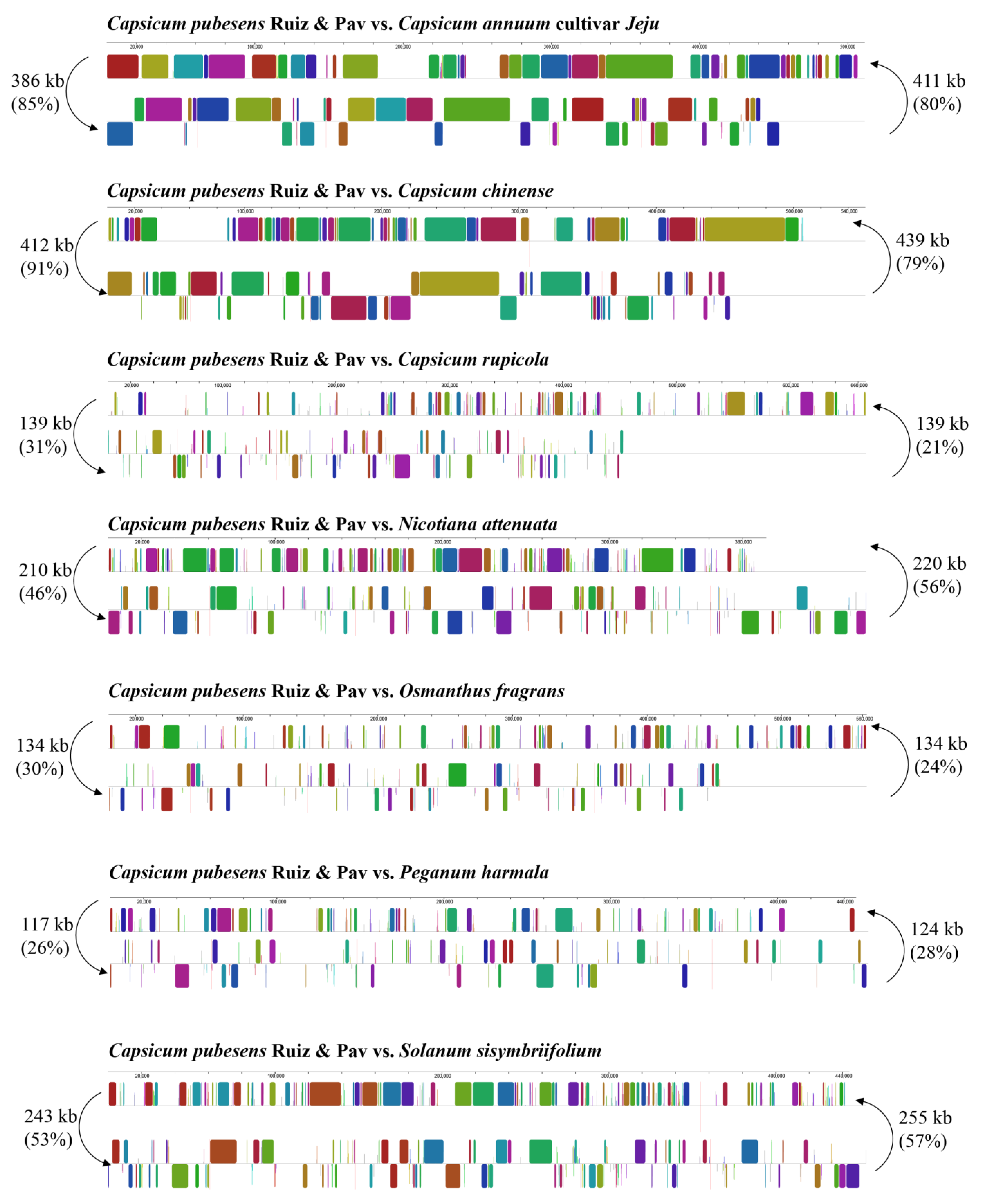
3.2. Competitive Analysis of C. pubescens Mt Genomes with Other Angiosperms
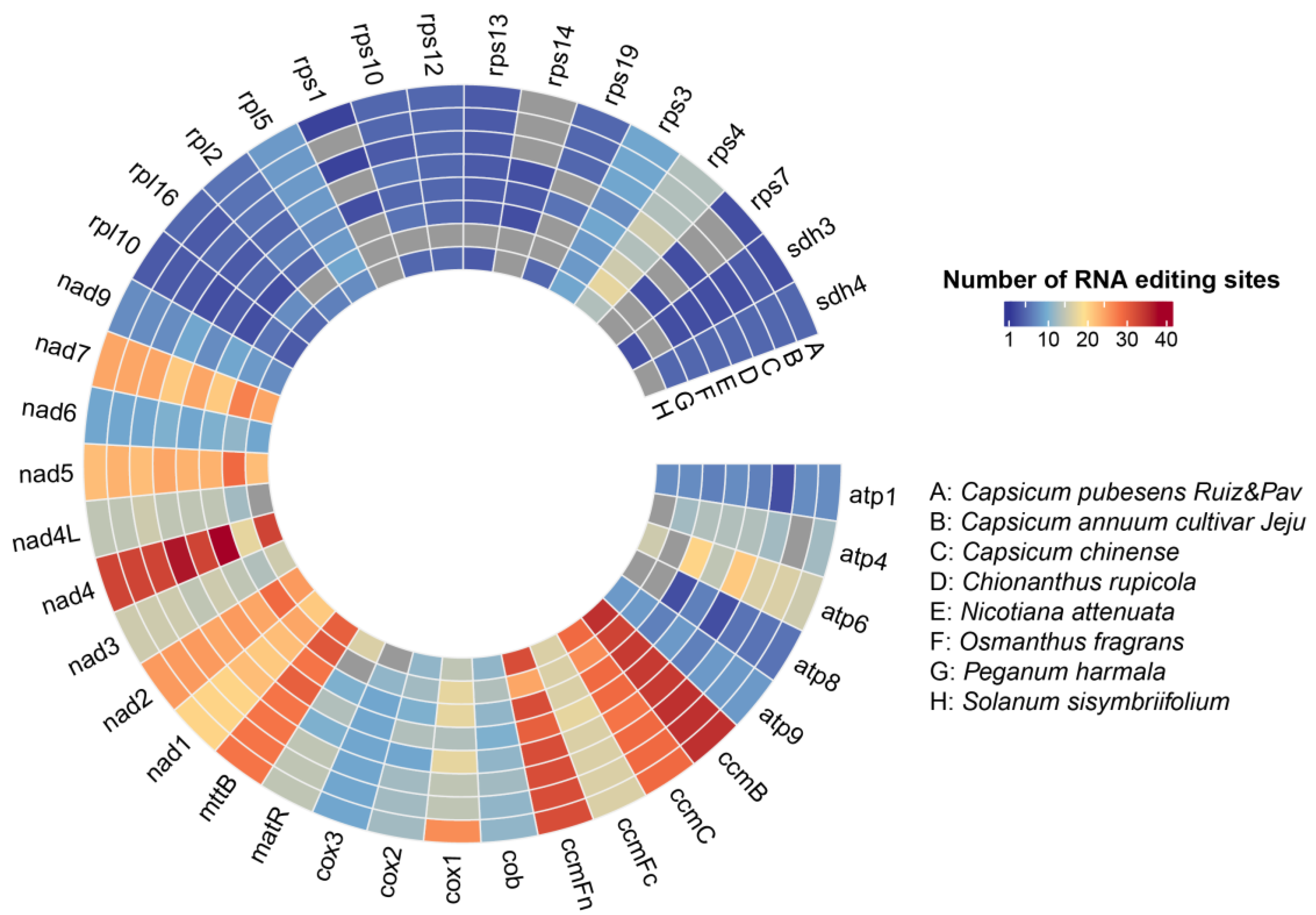
3.2.1. Codon Usage Analysis and RNA Editing Prediction in PCGs
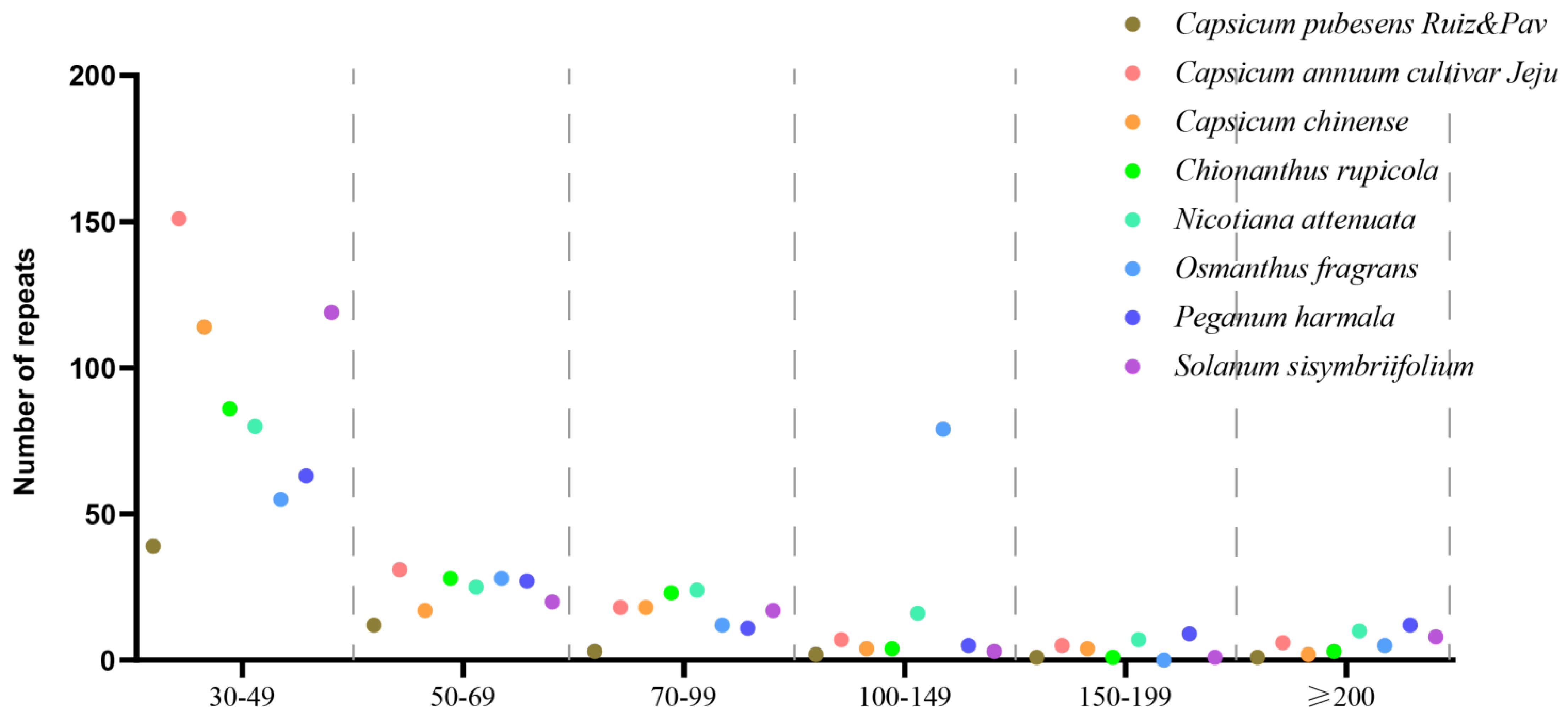
3.2.2. Dispersed Repeats Analysis
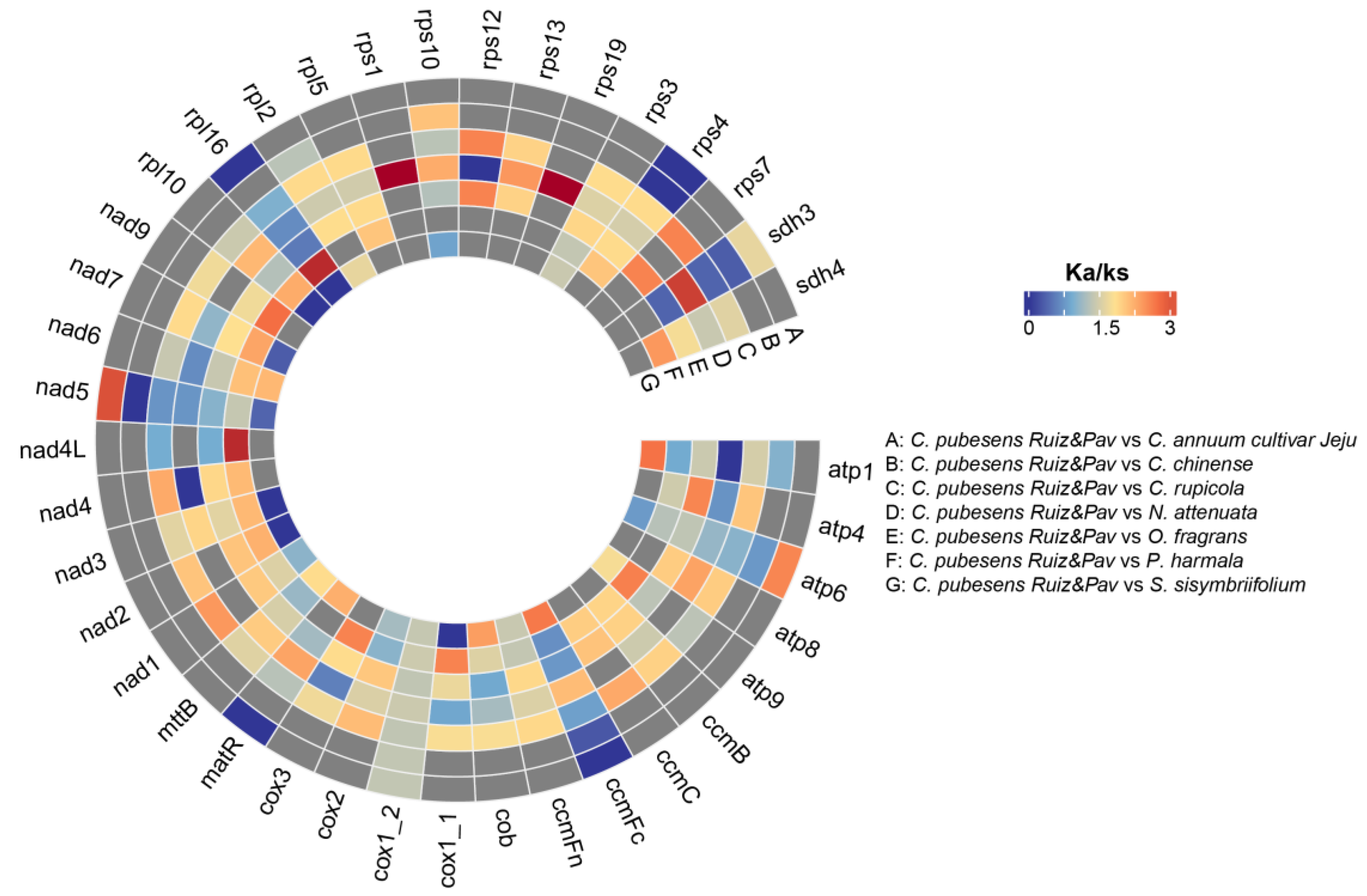
3.2.3. Selective Pressure Analysis
3.2.4. Gene Clusters and Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Costa, J.; Rodríguez, R.; Santos, C.; Soares, C.; Lima, N.; Santos, C. Mycobiota in Chilean chilli Capsicum annuum L. used for production of Merken. Int. J. Food. Microbiol. 2020, 334, 108833. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, D. The Chile Pepper Enclycopedia; HarperCollins: New York, NY, USA, 1999. [Google Scholar]
- Poyrazolu, E.S.; Yemiş, O.; Kadakal, Ç.; Artık, N. Determination of capsaicinoid profile of different chilli peppers grown in Turkey. J. Sci. Food Agric. 2005, 85, 1435–1438. [Google Scholar] [CrossRef]
- Namesny, A. Pimientos, 2nd ed.; Ediciones de Horticultura: Tarragona, Spain, 2006. [Google Scholar]
- DeWitt, D.; Bosland, P.W. Peppers of the World: An Identification Guide; Ten Speed Press: Berkeley, CA, USA, 1996. [Google Scholar]
- Rodriguez-Burruezo, A.; Prohens, J.; Raigon, M.D.; Nuez, F. Variation for bioactive compounds in aji (Capsicum baccatum L.) and rocoto (C-pubescens R. & P.) and implications for breeding. Euphytica 2009, 170, 169–181. [Google Scholar]
- Pickersgill, B. Genetic resources and breeding of Capsicum spp. Euphytica 1997, 96, 129–133. [Google Scholar] [CrossRef]
- Cecchhi, H.M.; Rodriguezamaya, D.B. Carotenoid composition and vitamin value of fresh and pasteurized cashew-apple (Anacardium occidentale L.) juice. J. Food Sci. 2010, 46, 147–149. [Google Scholar] [CrossRef]
- Bahat, A.; Gross, A. Mitochondrial plasticity in cell fate regulation. J. Biol. Chem. 2019, 294, 13852–13863. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, P.; Kannan, P.; Mlody, B.; Prigione, A. Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep. 2018, 19, e45432. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary mitochondrial disease and secondary mitochondrial dysfunction: Importance of distinction for diagnosis and treatment. Mol. Syndromol. 2016, 7, 122–137. [Google Scholar] [CrossRef]
- Knoop, V.; Volkmar, U.; Hecht, J.; Grewe, F. Mitochondrial genome evolution in the plant lineage. In Plant Mitochondria: Advances in Plant Biology; Springer: New York, NY, USA, 2011; pp. 3–29. [Google Scholar]
- Alverson, A.J.; Wei, X.; Rice, D.W.; Stern, D.B.; Barry, K.; Palmer, J.D. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 2010, 27, 1436–1448. [Google Scholar] [CrossRef]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and recombination of the bacterial sized Mult chromosomal mitochondrial genome of cucumber. Plant Cell 2011, 23, 2499–2513. [Google Scholar] [CrossRef]
- Bock, R. Witnessing genome evolution: Experimental reconstruction of endosymbiotic and horizontal gene transfer. Annu. Rev. Genet. 2017, 51, 1–22. [Google Scholar] [CrossRef]
- Rice, D.W.; Alverson, A.J.; Richardson, A.O.; Young, G.J.; Sanchez-Puerta, M.V.; Munzinger, J.; Barry, K.; Boore, J.L.; Zhang, Y.; Depamphilis, C.W.; et al. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 2013, 342, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comp. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed]
- Correspondence Analysis of Codon Usage. Available online: http://codonw.sourceforge.net/ (accessed on 19 December 2020).
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinf. 2010, 8, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. PREP-Mt: Predictive RNA editor for plant mitochondrial genes. BMC Bioinform. 2005, 6, 96. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids. Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Tian, X.; Zheng, J.; Hu, S.; Yu, J. The rice mitochondrial genomes and their variations. Plant Physiol. 2006, 140, 401–410. [Google Scholar] [CrossRef]
- Wang, P.; Lu, Q.; Ai, Y.; Wang, Y.; Li, T.; Wu, L.; Liu, J.; Cheng, Q.; Sun, L.; Shen, H. Candidate Gene Selection for Cytoplasmic Male Sterility in Pepper (Capsicum annuum L.) through Whole Mitochondrial Genome Sequencing. Int. J. Mol. Sci. 2019, 20, 578. [Google Scholar] [CrossRef]
- Jo, Y.D.; Choi, Y.; Kim, D.H.; Kim, B.D.; Kang, B.C. Extensive structural variations between mitochondrial genomes of CMS and normal peppers (Capsicum annuum L.) revealed by complete nucleotide sequencing. BMC Genom. 2014, 15, 561. [Google Scholar] [CrossRef]
- Cho, K.S.; Cho, J.H.; Im, J.S.; Choi, J.G.; Park, Y.E.; Hong, S.Y.; Kwon, M.; Kang, J.H.; Park, T.H. The complete mitochondrial genome sequences of potato (Solanum tuberosum L., Solanaceae). Mitochondrial DNA B Resour. 2017, 2, 781–782. [Google Scholar] [CrossRef]
- Bi, C.; Lu, N.; Xu, Y.; He, C.; Lu, Z. Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by comparative genomic approaches. Int. J. Mol. Sci. 2020, 21, 3778. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Genes | Gene Name | ||||
|---|---|---|---|---|---|
| cir 1 | cir 2 | cir 3 | cir 4 | Numbers | |
| Complex I (NADH dehydrogenase) | nad1 #, nad4 | - | nad1 #, nad2 #, nad3, nad4L, nad5 #, nad7, nad9 | nad1 #, nad2 #, nad5 #, nad6 | 9 |
| Complex II (succinate dehydrogenase) | - | - | sdh4 | sdh3 | 2 |
| Complex III (ubiquinol cytochrome c reductase) | - | cob | - | - | 1 |
| Complex IV (cytochrome c oxidase) | cox1 | - | cox1, cox3 | cox2 | 4 |
| Complex V (ATP synthase) | atp6 | atp9 | atp1, atp4, atp8 | - | 5 |
| Cytochrome c biogenesis | - | ccmFn | ccmB, ccmFc | ccmC | 4 |
| Ribosomal protein | rps7, rps10 | rpl5 | rps1, rps4, rps12, rps13, rpl2, rpl10 | rps3, rps19, rpl16 | 12 |
| Ribosomal RNA | - | rns, rrn5 | rnl | - | 3 |
| Transfer RNA | trnF, trnM, trnP, trnR, trnS | trnD, trnS | trnC, trnD, trnE(2) *, trnG, trnH, trnI, trnK, trnL, trnM(2) *, trnN, trnP, trnQ, trnT, trnW, trnY(2) * | trnL(2) *, trnM | 28 |
| Others | - | mttB | - | matR | 2 |
| Total | 70 | 70 | |||
| Length of Repeats | C. pubescens Ruiz & Pav | C. annuum cultivar Jeju | C. chinense | C. rupicola | N. attenuata | O. fragrans | P. harmala | S. sisymbriifolium |
|---|---|---|---|---|---|---|---|---|
| 30–49 | 39 | 151 | 114 | 86 | 80 | 55 | 63 | 119 |
| 50–69 | 12 | 31 | 17 | 28 | 25 | 28 | 27 | 20 |
| 70–99 | 3 | 18 | 18 | 23 | 24 | 12 | 11 | 17 |
| 100–149 | 2 | 7 | 4 | 4 | 16 | 79 | 5 | 3 |
| 150–199 | 1 | 5 | 4 | 1 | 7 | 0 | 9 | 1 |
| ≥200 | 1 | 6 | 2 | 3 | 10 | 5 | 12 | 8 |
| Total | 58 | 218 | 159 | 145 | 162 | 179 | 127 | 168 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, D.; Fu, W.; Fan, G.; Huang, D.; Wu, K.; Zhan, Y.; Tu, X.; He, J. Characteristics and Comparative Analysis of the Special-Structure (Non-Single-Circle) Mitochondrial Genome of Capsicum pubescens Ruiz & Pav. Genes 2024, 15, 152. https://doi.org/10.3390/genes15020152
Wu D, Fu W, Fan G, Huang D, Wu K, Zhan Y, Tu X, He J. Characteristics and Comparative Analysis of the Special-Structure (Non-Single-Circle) Mitochondrial Genome of Capsicum pubescens Ruiz & Pav. Genes. 2024; 15(2):152. https://doi.org/10.3390/genes15020152
Chicago/Turabian StyleWu, Di, Wenting Fu, Gaoling Fan, Dongfu Huang, Kangyun Wu, Yongfa Zhan, Xiangmin Tu, and Jianwen He. 2024. "Characteristics and Comparative Analysis of the Special-Structure (Non-Single-Circle) Mitochondrial Genome of Capsicum pubescens Ruiz & Pav" Genes 15, no. 2: 152. https://doi.org/10.3390/genes15020152
APA StyleWu, D., Fu, W., Fan, G., Huang, D., Wu, K., Zhan, Y., Tu, X., & He, J. (2024). Characteristics and Comparative Analysis of the Special-Structure (Non-Single-Circle) Mitochondrial Genome of Capsicum pubescens Ruiz & Pav. Genes, 15(2), 152. https://doi.org/10.3390/genes15020152