
The Impact of Bevacizumab and miR200c on EMT and EGFR-TKI Resistance in EGFR-Mutant Lung Cancer Organoids
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. The Construction of Human Lung Cancer Spheroids and Organoids
2.3. Detection of Epithelial–Mesenchymal Transition (EMT)
2.4. Mimicking Micro-RNA200c (miR200c)
2.5. Total RNA Isolation, Reverse-Transcription and Polymerase Chain Reaction (PCR)
2.6. Immunohistochemical Analyses
2.7. Cell Viability
2.8. Immunofluorescence Analyses
2.9. Western Blotting
2.10. Statistical Analysis
3. Results
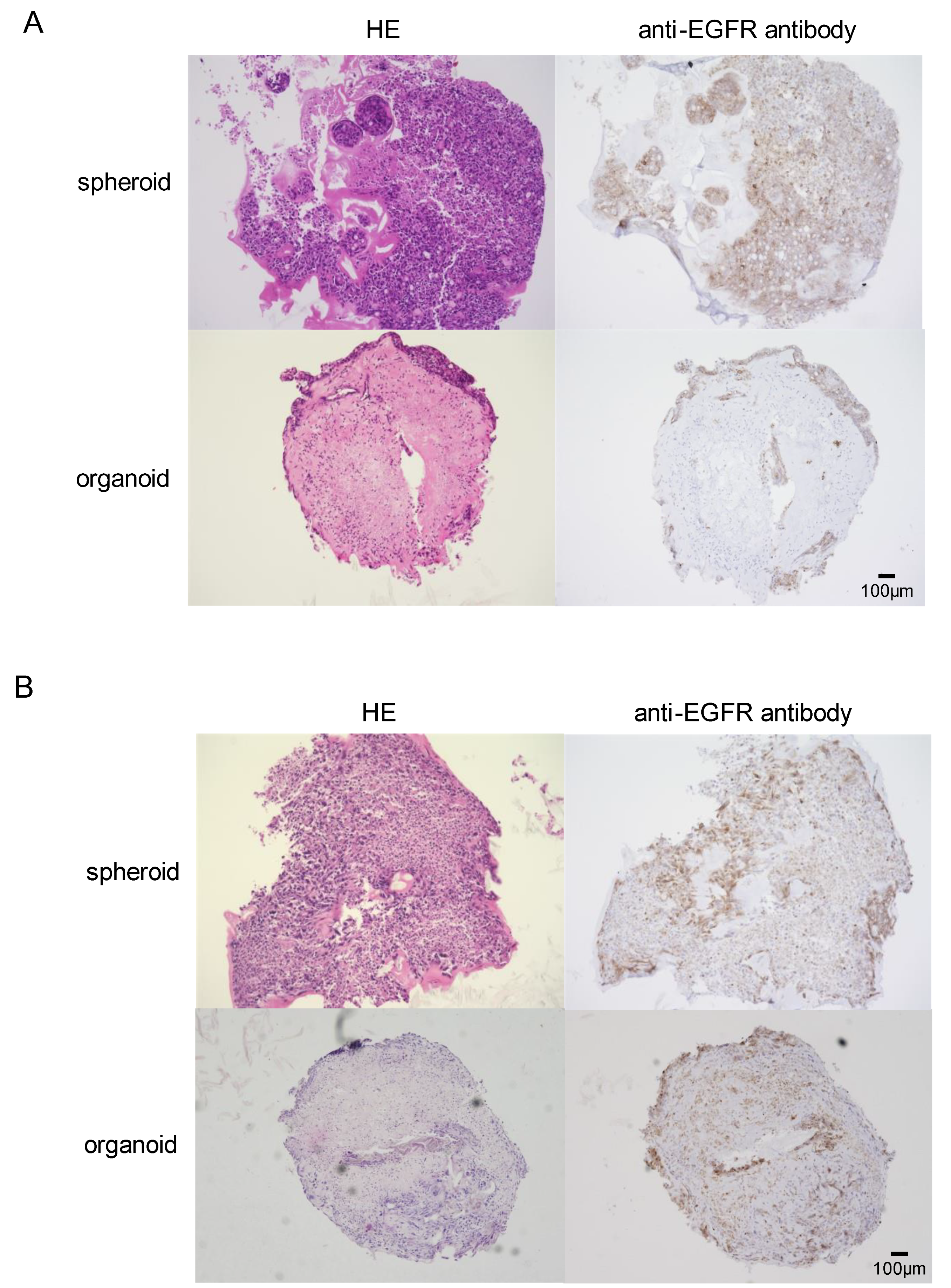
3.1. In Vitro Development of EGFR-Mutant Lung Cancer Organoids
3.2. Differential Susceptibility of Spheroids and Organoids to EGFR-TKI
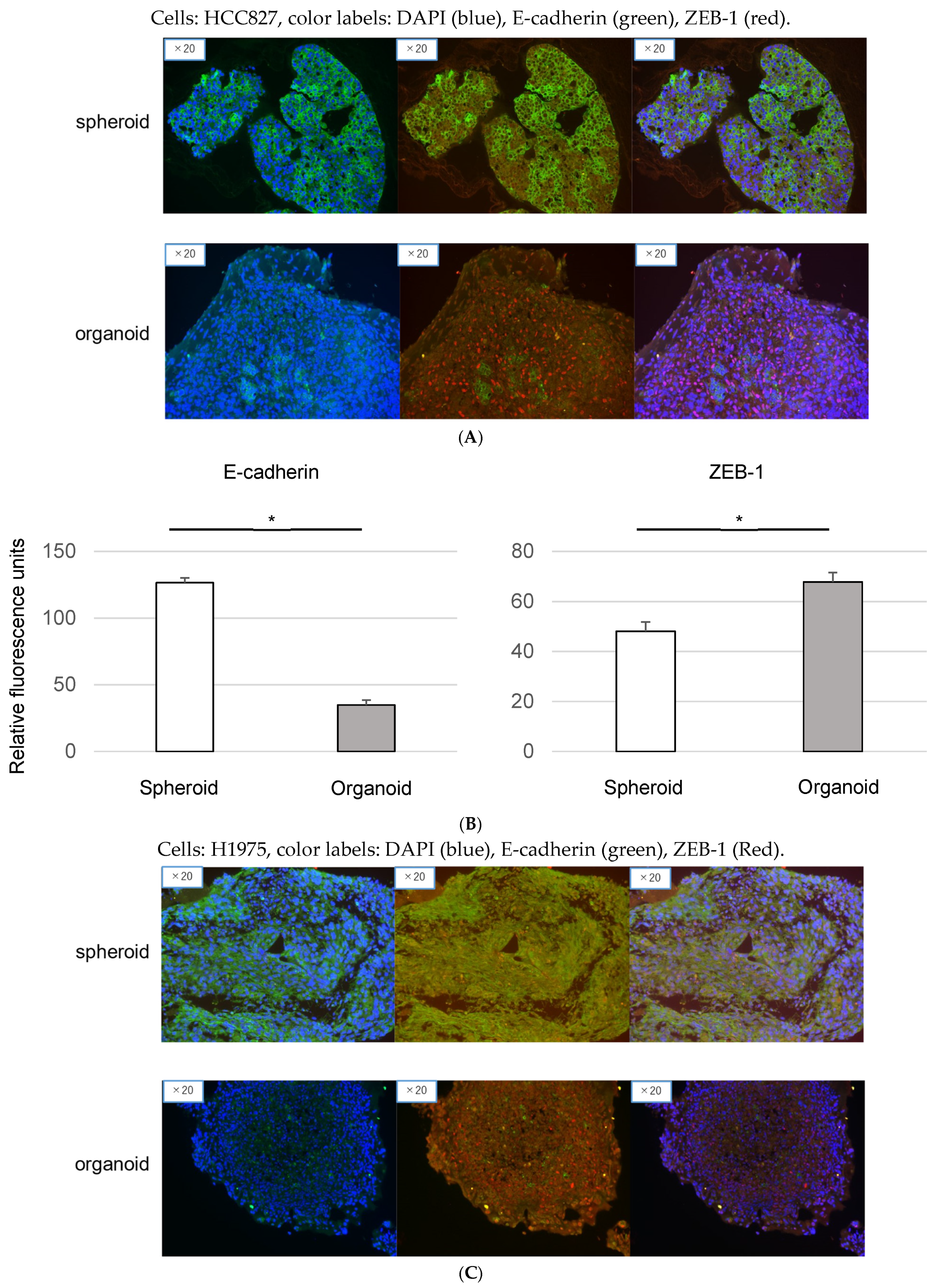
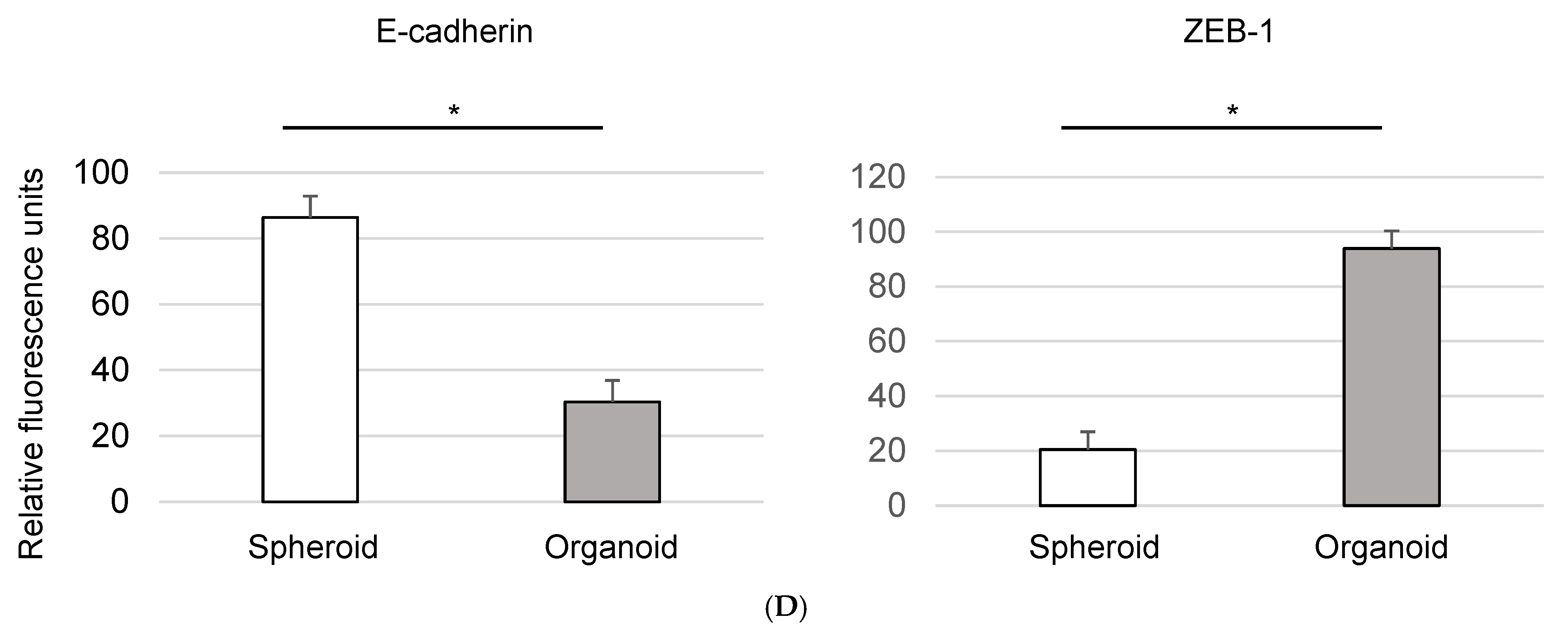
3.3. Epithelial-to-Mesenchymal Transition in Organoids: Immunofluorescence Analysis of EMT Markers
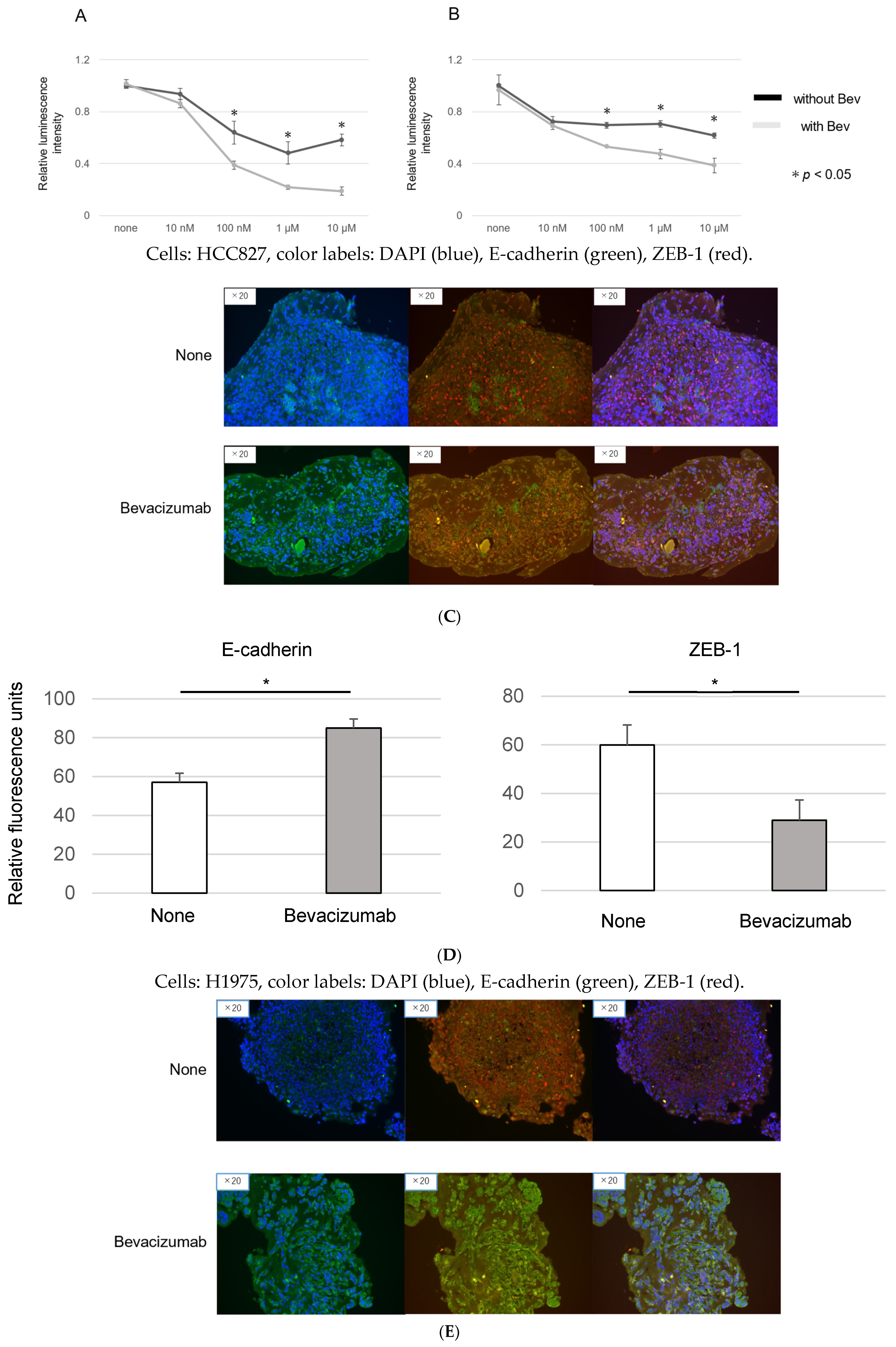
3.4. Modulation of EMT and EGFR-TKI Resistance in Organoids by Bevacizumab
3.5. Bevacizumab Inhibits on EMT in Cancer Organoids
3.6. miR200c Modulates EMT and Enhances EGFR-TKI Sensitivity in Organoids
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Burden of Disease Cancer Collaboration. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [CrossRef]
- Shi, Y.; Au, J.S.-K.; Thongprasert, S.; Srinivasan, S.; Tsai, C.-M.; Khoa, M.T.; Heeroma, K.; Itoh, Y.; Cornelio, G.; Yang, P.-C. A Prospective, Molecular Epidemiology Study of EGFR Mutations in Asian Patients with Advanced Non–Small-Cell Lung Cancer of Adenocarcinoma Histology (PIONEER). J. Thorac. Oncol. 2014, 9, 154–162. [Google Scholar] [CrossRef]
- Lee, D.H. Treatments for EGFR-mutant non-small cell lung cancer (NSCLC): The road to a success, paved with failures. Pharmacol. Ther. 2017, 174, 1–21. [Google Scholar] [CrossRef]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Noronha, A.; Nataraj, N.B.; Lee, J.S.; Zhitomirsky, B.; Oren, Y.; Oster, S.; Lindzen, M.; Mukherjee, S.; Will, R.; Ghosh, S.; et al. AXL and Error-Prone DNA Replication Confer Drug Resistance and Offer Strategies to Treat EGFR-Mutant Lung Cancer. Cancer Discov. 2022, 12, 2666–2683. [Google Scholar] [CrossRef]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin. Ther. Targets 2010, 14, 1073–1090. [Google Scholar] [CrossRef]
- Pool, M.; de Boer, H.R.; Hooge, M.N.L.-D.; van Vugt, M.A.; de Vries, E.G. Harnessing Integrative Omics to Facilitate Molecular Imaging of the Human Epidermal Growth Factor Receptor Family for Precision Medicine. Theranostics 2017, 7, 2111–2133. [Google Scholar] [CrossRef]
- Kobayashi, N.; Katakura, S.; Kamimaki, C.; Somekawa, K.; Fukuda, N.; Tanaka, K.; Watanabe, K.; Horita, N.; Hara, Y.; Piao, H.; et al. Resistance mechanisms of epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer patients: A meta-analysis. Thorac. Cancer 2021, 12, 1096–1105. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Han, J.-Y.; Ahn, M.-J.; Cho, B.C.; Yu, H.; Kim, S.-W.; Yang, J.C.-H.; Lee, J.S.; Su, W.-C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Tulchinsky, E.; Demidov, O.; Kriajevska, M.; Barlev, N.A.; Imyanitov, E. EMT: A mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2019, 1871, 29–39. [Google Scholar] [CrossRef]
- Myers, S.H.; Brunton, V.G.; Unciti-Broceta, A. AXL Inhibitors in Cancer: A Medicinal Chemistry Perspective. J. Med. Chem. 2016, 59, 3593–3608. [Google Scholar] [CrossRef] [PubMed]
- Sasai, Y. Next-Generation Regenerative Medicine: Organogenesis from Stem Cells in 3D Culture. Cell Stem Cell 2013, 12, 520–530. [Google Scholar] [CrossRef]
- Ogawa, H.; Koyanagi-Aoi, M.; Otani, K.; Zen, Y.; Maniwa, Y.; Aoi, T. Interleukin-6 blockade attenuates lung cancer tissue construction integrated by cancer stem cells. Sci. Rep. 2017, 7, 12317. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef]
- Mutlu, M.; Raza, U.; Saatci, Ö.; Eyüpoğlu, E.; Yurdusev, E.; Sahin, Ö. miR-200c: A versatile watchdog in cancer progression, EMT, and drug resistance. J. Mol. Med. 2016, 94, 629–644. [Google Scholar] [CrossRef]
- Maione, P.; Sacco, P.C.; Sgambato, A.; Casaluce, F.; Rossi, A.; Gridelli, C. Overcoming resistance to targeted therapies in NSCLC: Current approaches and clinical application. Ther. Adv. Med Oncol. 2015, 7, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Cortot, A.B.; Jänne, P.A. Molecular mechanisms of resistance in epidermal growth factor receptor-mutant lung adenocarcinomas. Eur. Respir. Rev. 2014, 23, 356–366. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting Epithelial–Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef]
- Shi, K.; Wang, G.; Pei, J.; Zhang, J.; Wang, J.; Ouyang, L.; Wang, Y.; Li, W. Emerging strategies to overcome resistance to third-generation EGFR inhibitors. J. Hematol. Oncol. 2022, 15, 94. [Google Scholar] [CrossRef]
- Wu, S.-G.; Shih, J.-Y. Management of acquired resistance to EGFR TKI–targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018, 17, 38. [Google Scholar] [CrossRef]
- Liu, X.; He, M.; Li, L.; Wang, X.; Han, S.; Zhao, J.; Dong, Y.; Ahmad, M.; Li, L.; Zhang, X.; et al. EMT and Cancer Cell Stemness Associated With Chemotherapeutic Resistance in Esophageal Cancer. Front. Oncol. 2021, 11, 672222. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Dou, B.; Tan, H.; Feng, Y.; Wang, N.; Wang, D. Tumor microenvironment-driven non-cell-autonomous resistance to antineoplastic treatment. Mol. Cancer 2019, 18, 69. [Google Scholar] [CrossRef]
- Bergfeld, S.A.; DeClerck, Y.A. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev. 2010, 29, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Zeng, S.; Wang, Z.; Wu, M.; Ma, Y.; Ye, X.; Zhang, B.; Liu, H. Cancer-associated fibroblasts promote epithelial-mesenchymal transition and EGFR-TKI resistance of non-small cell lung cancers via HGF/IGF-1/ANXA2 signaling. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Bremnes, R.M.; Veve, R.; Hirsch, F.R.; Franklin, W.A. The E-cadherin cell–cell adhesion complex and lung cancer invasion, metastasis, and prognosis. Lung Cancer 2002, 36, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Qiang, H.; Chang, Q.; Xu, J.; Qian, J.; Zhang, Y.; Lei, Y.; Han, B.; Chu, T. New advances in antiangiogenic combination therapeutic strategies for advanced non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Fukuhara, T.; Furuya, N.; Watanabe, K.; Sugawara, S.; Iwasawa, S.; Tsunezuka, Y.; Yamaguchi, O.; Okada, M.; Yoshimori, K.; et al. Erlotinib plus bevacizumab versus erlotinib alone in patients with EGFR-positive advanced non-squamous non-small-cell lung cancer (NEJ026): Interim analysis of an open-label, randomised, multicentre, phase 3 trial. Lancet Oncol. 2019, 20, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lin, G.; Chen, K.; Liang, R.; Wan, F.; Zhang, C.; Tian, G.; Zhu, X. VEGF promotes migration and invasion by regulating EMT and MMPs in nasopharyngeal carcinoma. J. Cancer 2020, 11, 7291–7301. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Choi, S.; Lee, W.-S. Bevacizumab and anexelekto inhibitor, TP-0903 inhibits TGF-β1-induced epithelial–mesenchymal transition of colon cancer cells. Anti-Cancer Drugs 2022, 33, e453–e461. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, C.; Cui, M.; Niu, J.; Ding, W. Inhibition of Bevacizumab-induced Epithelial–Mesenchymal Transition by BATF2 Overexpression Involves the Suppression of Wnt/β-Catenin Signaling in Glioblastoma Cells. Anticancer. Res. 2017, 37, 4285–4294. [Google Scholar] [CrossRef] [PubMed]
- Humphries, B.; Yang, C. The microRNA-200 family: Small molecules with novel roles in cancer development, progression and therapy. Oncotarget 2015, 6, 6472–6498. [Google Scholar] [CrossRef] [PubMed]
- Shien, K.; Toyooka, S.; Yamamoto, H.; Soh, J.; Jida, M.; Thu, K.L.; Hashida, S.; Maki, Y.; Ichihara, E.; Asano, H.; et al. Acquired Resistance to EGFR Inhibitors Is Associated with a Manifestation of Stem Cell–like Properties in Cancer Cells. Cancer Res. 2013, 73, 3051–3061. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, N.; Katakura, S.; Fukuda, N.; Somekawa, K.; Kaneko, A.; Kaneko, T. The Impact of Bevacizumab and miR200c on EMT and EGFR-TKI Resistance in EGFR-Mutant Lung Cancer Organoids. Genes 2024, 15, 1624. https://doi.org/10.3390/genes15121624
Kobayashi N, Katakura S, Fukuda N, Somekawa K, Kaneko A, Kaneko T. The Impact of Bevacizumab and miR200c on EMT and EGFR-TKI Resistance in EGFR-Mutant Lung Cancer Organoids. Genes. 2024; 15(12):1624. https://doi.org/10.3390/genes15121624
Chicago/Turabian StyleKobayashi, Nobuaki, Seigo Katakura, Nobuhiko Fukuda, Kohei Somekawa, Ayami Kaneko, and Takeshi Kaneko. 2024. "The Impact of Bevacizumab and miR200c on EMT and EGFR-TKI Resistance in EGFR-Mutant Lung Cancer Organoids" Genes 15, no. 12: 1624. https://doi.org/10.3390/genes15121624
APA StyleKobayashi, N., Katakura, S., Fukuda, N., Somekawa, K., Kaneko, A., & Kaneko, T. (2024). The Impact of Bevacizumab and miR200c on EMT and EGFR-TKI Resistance in EGFR-Mutant Lung Cancer Organoids. Genes, 15(12), 1624. https://doi.org/10.3390/genes15121624