
Segregation of the COL6A2 Variant (c.1817-3C>G) in a Consanguineous Saudi Family with Bethlem Myopathy



Abstract
1. Introduction
2. Case Report
2.1. Extraction of Genomic DNA and Whole-Exome Sequencing
2.2. Ethical Consideration and Consent for Participation and Publication
3. Results
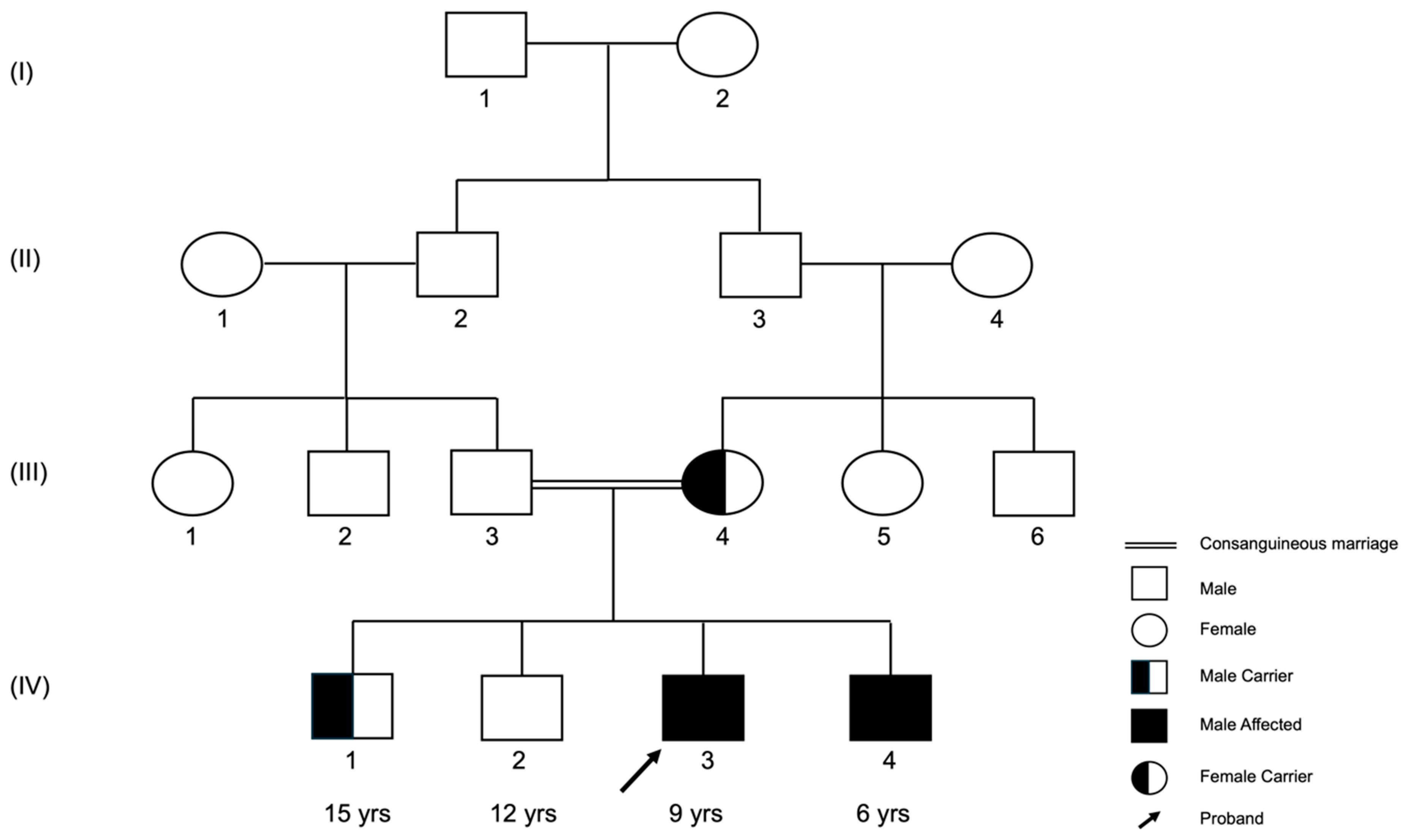
3.1. The (c.1817-3C>G) Variant Was Recessively Inherited in the Family
3.2. The (c.1817-3C>G) Variant Is Predicted to Be an Intronic and Likely Pathogenic

{kind=link}
{kind=link}
| SNV rs112645828 | SpliceAI | dbscSNV | FATHMM-MKL | MaxEntScan |
|---|---|---|---|---|
| Reported score | 0.650 | 0.998 | 0.991 | 4.677 |
| Interpretation | Acceptor loss | Pathogenic supporting | Pathogenic supporting | Pathogenic supporting |
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huml, R.A. Muscular Dystrophy: Historical Background and Types. In Muscular Dystrophy; Oxford University Press: Oxford, UK, 2015; pp. 5–7. [Google Scholar]
- Gualandi, F.; Urciuolo, A.; Martoni, E.; Sabatelli, P.; Squarzoni, S.; Bovolenta, M.; Messina, S.; Mercuri, E.; Franchella, A.; Ferlini, A.; et al. Autosomal Recessive Bethlem Myopathy. Neuromuscul. Disord. 2009, 19, 608–609. [Google Scholar] [CrossRef]
- Bönnemann, C.G. The Collagen VI-Related Myopathies. Handb. Clin. Neurol. 2011, 101, 81–96. [Google Scholar] [CrossRef]
- Norwood, F.L.M.; Harling, C.; Chinnery, P.F.; Eagle, M.; Bushby, K.; Straub, V. Prevalence of Genetic Muscle Disease in Northern England: In-Depth Analysis of a Muscle Clinic Population. Brain 2009, 132, 3175–3186. [Google Scholar] [CrossRef]
- Saroja, A.O.; Naik, K.R.; Nalini, A.; Gayathri, N. Bethlem Myopathy: An Autosomal Dominant Myopathy with Flexion Contractures, Keloids, and Follicular Hyperkeratosis. Ann. Indian Acad. Neurol. 2013, 16, 712–715. [Google Scholar] [CrossRef]
- Elmas, M.; Gogus, B. A Case of Bethlem Myopathy with Autosomal Recessive Inheritance with a Novel Mutation in the COL6A2 Gene. J. Biochem. Clin. Genet. 2018, 1, 81–83. [Google Scholar] [CrossRef]
- Shih, J.A.; Folch, A.; Wong, B.L. Duchenne Muscular Dystrophy: The Heart of the Matter. Curr. Heart Fail. Rep. 2020, 17, 57–66. [Google Scholar] [CrossRef]
- Galli, F.; Bragg, L.; Meggiolaro, L.; Rossi, M.; Caffarini, M.; Naz, N.; Santoleri, S.; Cossu, G. Gene and Cell Therapy for Muscular Dystrophies: Are We Getting There? Hum. Gene Ther. 2018, 29, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Alkuraya, F.S. Genetics and Genomic Medicine in Saudi Arabia. Mol. Genet. Genom. Med. 2014, 2, 369–378. [Google Scholar] [CrossRef]
- Bamaga, A.K.; Alghamdi, F.; Alshaikh, N.; Altwaijri, W.; Bashiri, F.A.; Hundallah, K.; Abukhaled, M.; Muthaffar, O.Y.; Al-Mehmadi, S.; Jamaly, T.A.; et al. Consensus Statement on the Management of Duchenne Muscular Dystrophy in Saudi Arabia During the Coronavirus Disease 2019 Pandemic. Front. Pediatr. 2021, 9, 629549. [Google Scholar] [CrossRef]
- Alghamdi, F.; Al-Tawari, A.; Alrohaif, H.; Alshuaibi, W.; Mansour, H.; Aartsma-Rus, A.; Mégarbané, A. Case Report: The Genetic Diagnosis of Duchenne Muscular Dystrophy in the Middle East. Front. Pediatr. 2021, 9, 716424. [Google Scholar] [CrossRef]
- O’Brien, M. Aids to the Examination of the Peripheral Nervous System: 6th Edition. Pract. Neurol. 2023, 23, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Gowers, W.R. Clinical Lecture ON PSEUDO-HYPERTROPHIC MUSCULAR PARALYSIS. Lancet 1879, 114, 73–75. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.; Boerwinkle, E.; Liu, X. In Silico Prediction of Splice-Altering Single Nucleotide Variants in the Human Genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum Entropy Modeling of Short Sequence Motifs with Applications to RNA Splicing Signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R.; Campbell, C. An Integrative Approach to Predicting the Functional Effects of Non-Coding and Coding Sequence Variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar; [VCV000974882.4]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000974882.4 (accessed on 5 September 2024).
- Lampe, A.K.; Dunn, D.M.; Von Niederhausern, A.C.; Hamil, C.; Aoyagi, A.; Laval, S.H.; Marie, S.K.; Chu, M.L.; Swoboda, K.; Muntoni, F.; et al. Automated Genomic Sequence Analysis of the Three Collagen VI Genes: Applications to Ullrich Congenital Muscular Dystrophy and Bethlem Myopathy. J. Med. Genet. 2005, 42, 108–120. [Google Scholar] [CrossRef]
- Rs112645828 (SNP)—Genes and Regulation—Homo_Sapiens—Ensembl Genome Browser 111. Available online: http://asia.ensembl.org/Homo_sapiens/Variation/Mappings?db=core;r=21:46124962-46125962;v=rs112645828;vdb=variation;vf=1079240881#ENST00000300527_1079240881_G_tablePanel (accessed on 9 May 2024).
- Ward, A.J.; Cooper, T.A. The Pathobiology of Splicing. J. Pathol. 2010, 220, 152–163. [Google Scholar] [CrossRef]
- Ward, L.D.; Kellis, M. Interpreting Noncoding Genetic Variation in Complex Traits and Human Disease. Nat. Biotechnol. 2012, 30, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; Cukura, A.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as Designed by Its Users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef]
- Zhang, R.Z.; Zou, Y.; Pan, T.C.; Markova, D.; Fertala, A.; Hu, Y.; Squarzoni, S.; Reed, U.C.; Marie, S.K.N.; Bönnemann, C.G.; et al. Recessive COL6A2 C-Globular Missense Mutations in Ullrich Congenital Muscular Dystrophy: Role of the C2a Splice Variant. J. Biol. Chem. 2010, 285, 10005–10015. [Google Scholar] [CrossRef]
- Full Data View for Gene COL6A2—Global Variome Shared LOVD. Available online: https://databases.lovd.nl/shared/view/COL6A2?search_VariantOnGenome%2FDBID=%3D%22COL6A2_000014%22 (accessed on 9 May 2024).
- 21-46125462-C-G|GnomAD v4.1.0|GnomAD. Available online: https://gnomad.broadinstitute.org/variant/21-46125462-C-G?dataset=gnomad_r4 (accessed on 9 May 2024).
- Wilpert, N.-M.; Schuelke, M.; Lala, B.; Weiß, C. A Mild But Typical Presentation of Bethlem Myopathy With a Novel In-Frame Deletion in COL6A1: Almost Overlooked. Neurology 2024, 102, e209476. [Google Scholar] [CrossRef]
- Bardakov, S.N.; Deev, R.V.; Magomedova, R.M.; Umakhanova, Z.R.; Allamand, V.; Gartioux, C.; Zulfugarov, K.Z.; Akhmedova, P.G.; Tsargush, V.A.; Titova, A.A.; et al. Intrafamilial Phenotypic Variability of Collagen VI-Related Myopathy Due to a New Mutation in the COL6A1 Gene. J. Neuromuscul. Dis. 2021, 8, 273–285. [Google Scholar] [CrossRef]
- Kachuei, M.; Orangi, K.; Mohammadi, A.; Mohammadi, M.; Mojbafan, M. Bethlem Myopathy: A Novel Homozygous Variant of c.385C>T (p.Arg129Cys) in the COL6A2 Gene. Clin. Case Rep. 2024, 12, e9306. [Google Scholar] [CrossRef]
- Baker, N.L.; Mörgelin, M.; Pace, R.A.; Peat, R.A.; Adams, N.E.; Gardner, R.J.M.K.; Rowland, L.P.; Miller, G.; De Jonghe, P.; Ceulemans, B.; et al. Molecular Consequences of Dominant Bethlem Myopathy Collagen VI Mutations. Ann. Neurol. 2007, 62, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Kutluk, M.G.; Kadem, N.; Bektas, O.; Randa, N.C.; Tuncer, G.O.; Albayrak, P.; Eminoglu, T.; Teber, S.T. Genotype-Phenotype Correlation of the Childhood-Onset Bethlem Myopathy in the Mediterranean Region of Turkey. Ann. Indian Acad. Neurol. 2021, 24, 547–551. [Google Scholar] [CrossRef]
- Oros, M.; Baranga, L.; Glangher, A.; Adina-Diana, M.; Jugulete, G.; Pavelescu, C.; Mihaltan, F.; Plaiasu, V.; Gheorghe, D.C. A Diagnostic Challenge in an Adolescent with Collagen VI-Related Myopathy and Emotional Disorder-Case Report. J. Pers. Med. 2023, 13, 1577. [Google Scholar] [CrossRef]
- Stavusis, J.; Micule, I.; Wright, N.T.; Straub, V.; Topf, A.; Panadés-de Oliveira, L.; Domínguez-González, C.; Inashkina, I.; Kidere, D.; Chrestian, N.; et al. Collagen VI-Related Limb-Girdle Syndrome Caused by Frequent Mutation in COL6A3 Gene with Conflicting Reports of Pathogenicity. Neuromuscul. Disord. 2020, 30, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.Y.; Qu, Y.J.; Song, F.; Sun, X.F.; Ge, X.S.; Jiao, H. [Clinical manifestations and genetics analysis of collagen type Ⅵ-related myopathy caused by variants in COL6A3 gene]. Zhonghua Er Ke Za Zhi 2019, 57, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Marakhonov, A.V.; Tabakov, V.Y.; Zernov, N.V.; Dadali, E.L.; Sharkova, I.V.; Skoblov, M.Y. Two Novel COL6A3 Mutations Disrupt Extracellular Matrix Formation and Lead to Myopathy from Ullrich Congenital Muscular Dystrophy and Bethlem Myopathy Spectrum. Gene 2018, 672, 165–171. [Google Scholar] [CrossRef]
- Collins, J.; Foley, A.R.; Straub, V.; Bönnemann, C.G. Spontaneous Keloid Formation in Patients with Bethlem Myopathy. Neurology 2012, 79, 2158. [Google Scholar] [CrossRef]
- Witting, N.; Krag, T.; Werlauff, U.; Duno, M.; Oestergaard, S.T.; Dahlqvist, J.R.; Vissing, J. Collagen XII Myopathy with Rectus Femoris Atrophy and Collagen XII Retention in Fibroblasts. Muscle Nerve 2018, 57, 1026–1030. [Google Scholar] [CrossRef]
- Punetha, J.; Kesari, A.; Hoffman, E.P.; Gos, M.; Kamińska, A.; Kostera-Pruszczyk, A.; Hausmanowa-Petrusewicz, I.; Hu, Y.; Zou, Y.; Bönnemann, C.G.; et al. Novel Col12A1 Variant Expands the Clinical Picture of Congenital Myopathies with Extracellular Matrix Defects. Muscle Nerve 2017, 55, 277–281. [Google Scholar] [CrossRef]
- Panadés-de Oliveira, L.; Rodríguez-López, C.; Cantero Montenegro, D.; Marcos Toledano, M.d.M.; Fernández-Marmiesse, A.; Esteban Pérez, J.; Hernández Lain, A.; Domínguez-González, C. Bethlem Myopathy: A Series of 16 Patients and Description of Seven New Associated Mutations. J. Neurol. 2019, 266, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.I.; Marques, C.; Pinto-Basto, J.; Negrão, L. Bethlem Myopathy in a Portuguese Patient—Case Report. Acta Myol. 2017, 36, 178–181. [Google Scholar]
- Pino, M.G.; Rich, K.A.; Kolb, S.J. Update on Biomarkers in Spinal Muscular Atrophy. Biomark Insights 2021, 16, 11772719211035643. [Google Scholar] [CrossRef]
- El Mouzan, M.I.; Al Salloum, A.A.; Al Herbish, A.S.; Qurachi, M.M.; Al Omar, A.A. Consanguinity and Major Genetic Disorders in Saudi Children: A Community-Based Cross-Sectional Study. Ann. Saudi Med. 2008, 28, 169–173. [Google Scholar] [CrossRef]
- El-Mouzan, M.I.; Al-Salloum, A.A.; Al-Herbish, A.S.; Qurachi, M.M.; Al-Omar, A.A. Regional Variations in the Prevalence of Consanguinity in Saudi Arabia. Saudi Med. J. 2007, 28, 1881–1884. [Google Scholar] [PubMed]
- Alkuraya, F.S. Discovery of Rare Homozygous Mutations from Studies of Consanguineous Pedigrees. Curr. Protoc. Hum. Genet. 2012, 75, 6.12.1–6.12.13. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing Technologies—The next Generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Merlini, L.; Morandi, L.; Granata, C.; Ballestrazzi, A. Bethlem Myopathy: Early-Onset Benign Autosomal Dominant Myopathy with Contractures. Description of Two New Families. Neuromuscul. Disord. 1994, 4, 503–511. [Google Scholar] [CrossRef]

| Case | Gene | Variant | Type | Genotype | Lost Ambulation | Walking Abnormality | Respiratory Symptoms | Muscle Weakness and Contractures |
|---|---|---|---|---|---|---|---|---|
| [31] (Wilpert et al., 2024) | COL6A1 | c.117_119delCCT | In-Frame deletion | Hh | Not reported | Truncal pendulum movements during walking. | Not reported | Elbow and long finger flexor contractures. Mild proximal muscle weakness |
| [32] (Bardakov et al., 2021) | COL6A1 | c.227 + 2T>C | Splice site | HH | Not reported | Not reported | Not reported | Early-onset severe proximal joint contractures and distal joint hypermobility |
| [33] (Kachuei et al., 2024) | COL6A2 | c.385C>T | Missense | HH | yes | Waddling gait and | Not reported | Proximal lower limb weakness, a positive Gowers’ sign, lumbar hyperlordosis and absent myotatic reflexes |
| [33] (Baker et al., 2007) | COL6A2 | c.1000-2A>G | Splice site | Hh | Not reported | Not reported | Not reported | Proximal lower limb muscle weakness and contractures of elbow, knee, and ankles |
| [6] (Elmas and Gogus, 2018) | COL6A2 | c.2584C>T (p.Arg862Trp) | Missense | Hh | Not reported | Not reported | Not reported | Joint contractures, finger flexors, neuromotor developmental delay |
| [34] (Kutluk et al., 2021) | COL6A2 | c.2096G>A | Missense | Hh | Not reported | Not reported | Not reported | Predominant proximal muscle weakness, contractures at metacarpals, distal hyperextensibility of fingers |
| [34] (Kutluk et al., 2021) | COL6A2 | c.2096G>A | Missense | Hh | Not reported | Not reported | Not reported | Predominant proximal muscle weakness, distal hyperextensibility of fingers |
| [35] (Oros et al., 2023) | COL6A2 | c.784G>T | Missense | Hh | Not reported | Difficulty to walk independently and running | Not reported | Progressive motor deficit involving all four limbs |
| [2] (Gualandi et al., 2009) | COL6A2 | Q819X; R366X | Missense | HH | Not reported | Not reported | Not reported | Proximal lower limb muscle weakness and contractures of fingers flexors muscles |
| [36] (Stavusis et al., 2020) | COL6A3 | c.7447A>G | Missense | Hh | Not reported | Not reported | Not reported | Tendon retractions, kyphoscoliosis |
| [37] (Peng et al., 2019) | COL6A3 | c.6229G>C, c.5169_5177del | Missense, Deletion | Hh | Not reported | Not reported | Not reported | Mild muscle weakness |
| [38] (Marakhonov et al., 2018) | COL6A3 | p.Glu2402Ter | Nonsense | HH | Not reported | Not reported | Not reported | Diffuse muscle weakness, striking distal joint hyperlaxity, proximal contractures, calcaneal protrusion, kyphosis, hip dislocation. |
| [39] (Collins et al., 2012) | COL6A3, COL6A2 | c.G6517T, c.G1861A | Missense | Hh | Not reported | Not reported | Not reported | Distal hyperlaxity (finger and ankles), proximal contractures (hips and knees), prominent calcaneus. |
| [40] (Witting et al., 2018) | COL12A1 | c.8100 + 2T>C | Missense | Hh | Not reported | Not reported | Not reported | Congenital hip dysplasia |
| [41] (Punetha et al., 2017) | COL12A1 | c.8329G>C | Missense | Hh | Not reported | Not reported | Not reported | Upper and lower limbs contractures followed by resolution of contractures, limited motor performance. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldharee, H.; Hamdan, H.Z. Segregation of the COL6A2 Variant (c.1817-3C>G) in a Consanguineous Saudi Family with Bethlem Myopathy. Genes 2024, 15, 1405. https://doi.org/10.3390/genes15111405
Aldharee H, Hamdan HZ. Segregation of the COL6A2 Variant (c.1817-3C>G) in a Consanguineous Saudi Family with Bethlem Myopathy. Genes. 2024; 15(11):1405. https://doi.org/10.3390/genes15111405
Chicago/Turabian StyleAldharee, Hitham, and Hamdan Z. Hamdan. 2024. "Segregation of the COL6A2 Variant (c.1817-3C>G) in a Consanguineous Saudi Family with Bethlem Myopathy" Genes 15, no. 11: 1405. https://doi.org/10.3390/genes15111405
APA StyleAldharee, H., & Hamdan, H. Z. (2024). Segregation of the COL6A2 Variant (c.1817-3C>G) in a Consanguineous Saudi Family with Bethlem Myopathy. Genes, 15(11), 1405. https://doi.org/10.3390/genes15111405