
The Relationship of Duffy Gene Polymorphism with High-Sensitivity C-Reactive Protein, Mortality, and Cardiovascular Outcomes in Black Individuals

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Methods
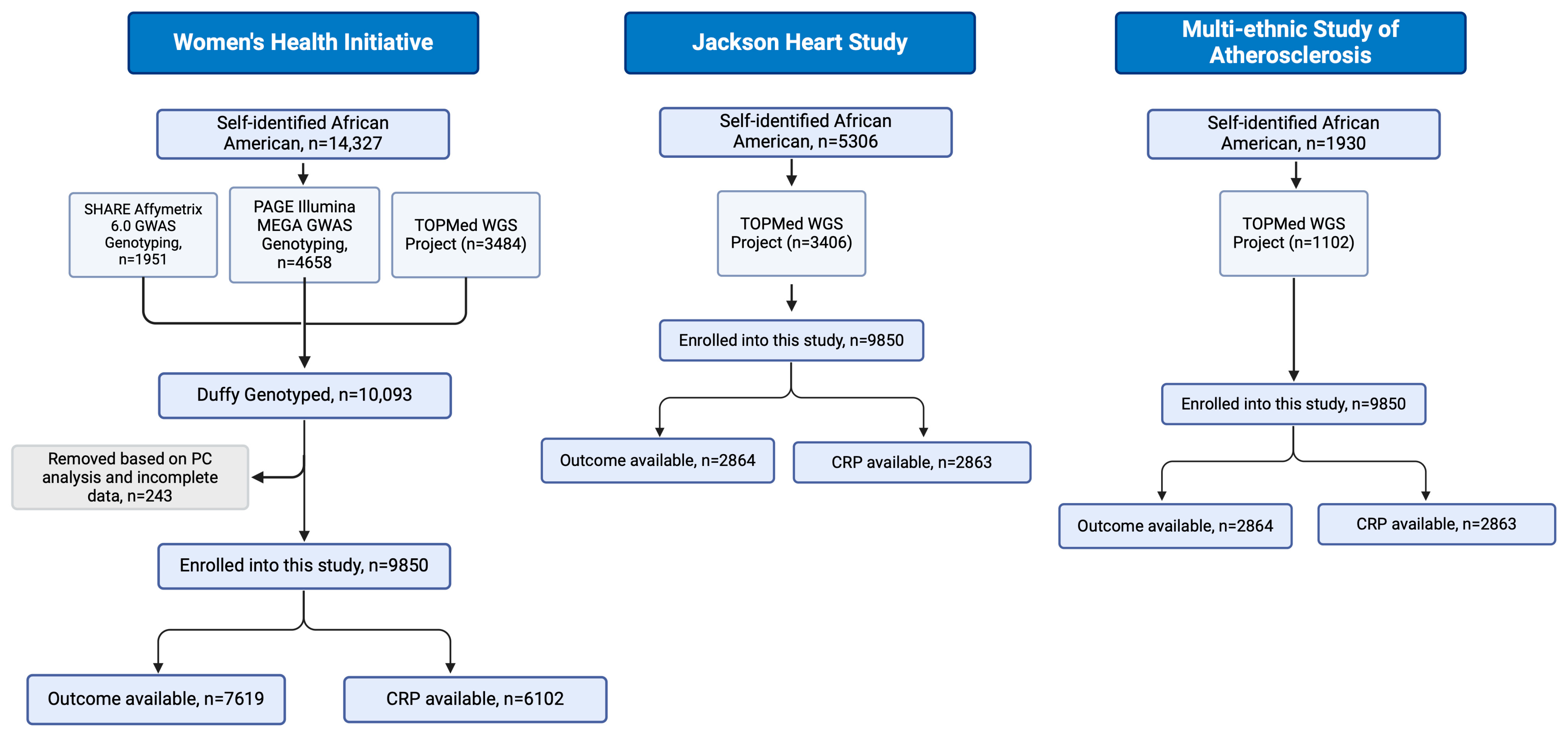
2.1. Study Design and Data Collection
2.2. Genotype Assessment of Duffy-Null Status, CRP Locus, and Principal Components of Genetic Ancestry
2.3. Laboratory Measurements of hs-CRP
2.4. Adjudication of Clinical Outcomes
2.5. Statistical Analysis
2.6. Replication Analysis in United Kingdom Biobank Repository
3. Results
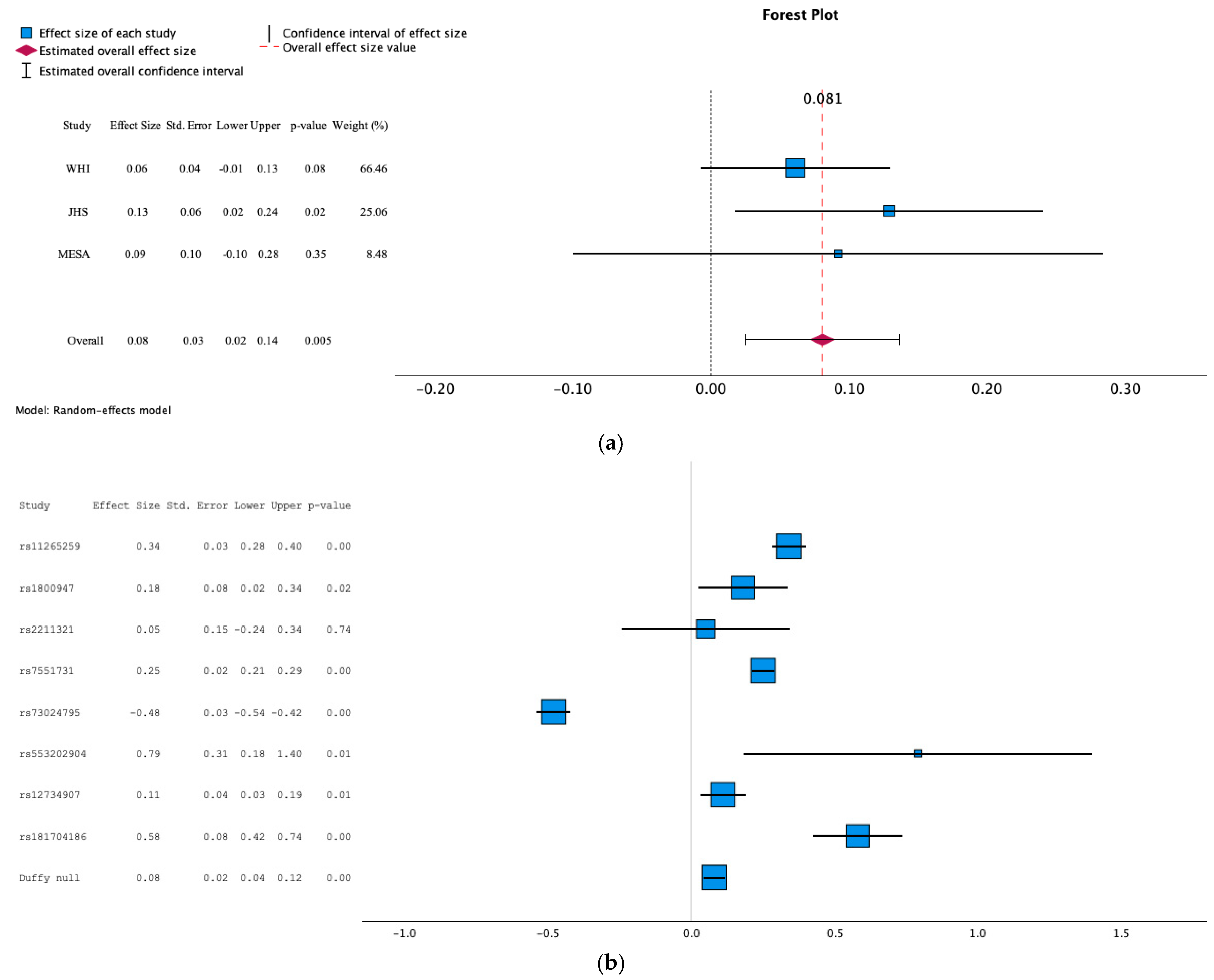
3.1. Association of Duffy Null with hs-CRP Levels
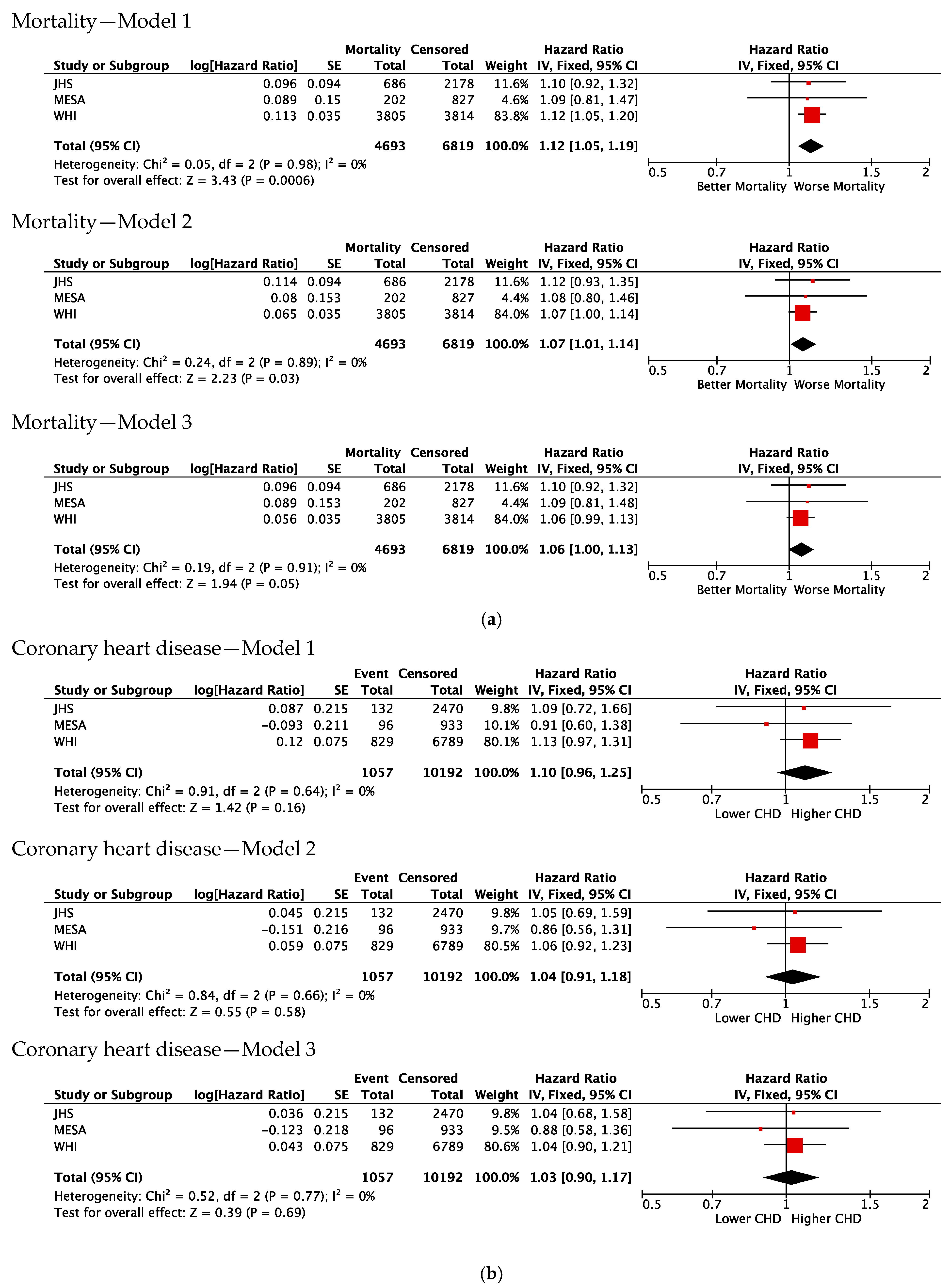
3.2. Association of Duffy Null with Risk of Mortality
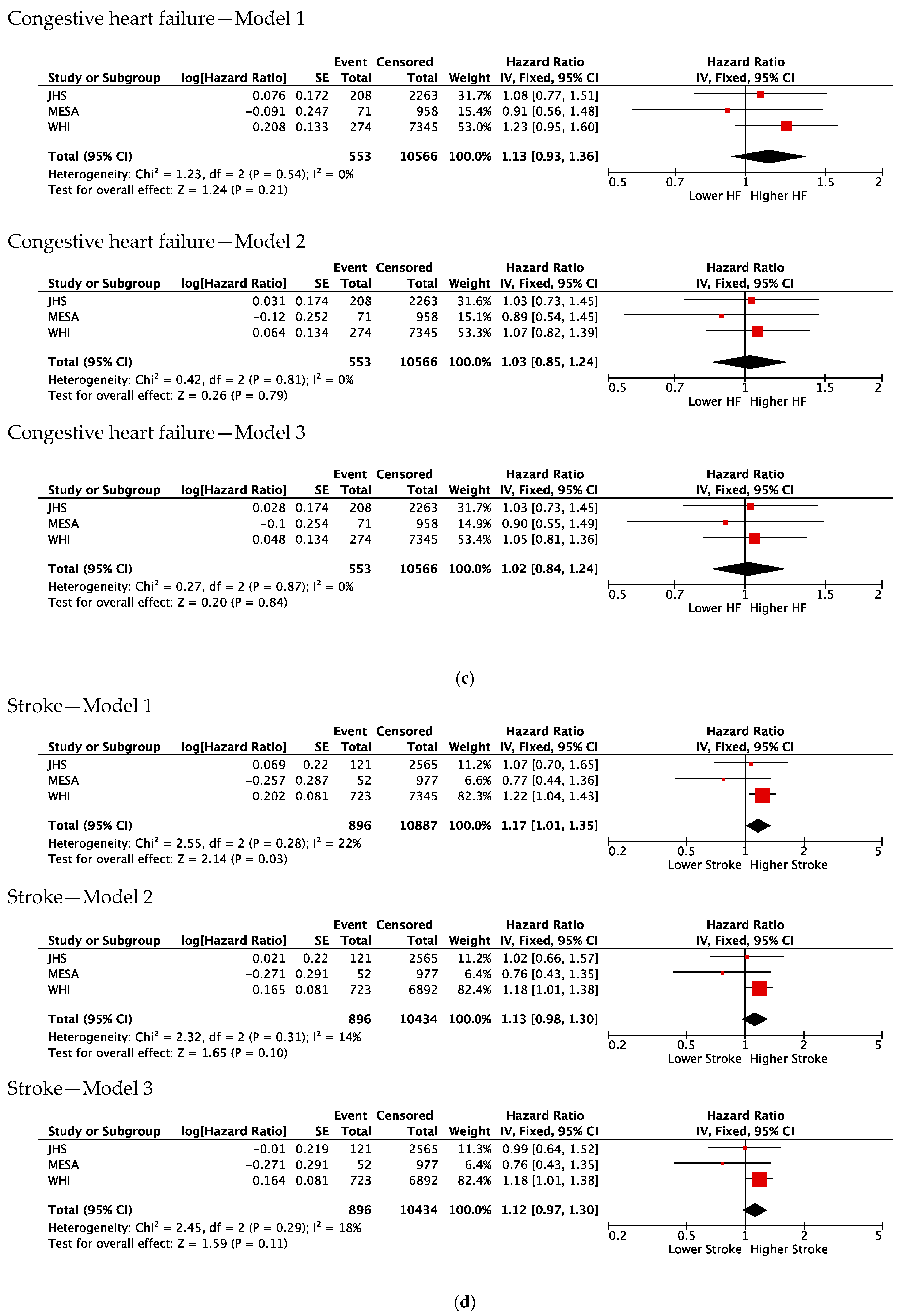
3.3. Association of Duffy Null with Incident CVD
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fujishiro, K.; Hajat, A.; Landsbergis, P.A.; Meyer, J.D.; Schreiner, P.J.; Kaufman, J.D. Explaining racial/ethnic differences in all-cause mortality in the Multi-Ethnic Study of Atherosclerosis (MESA): Substantive complexity and hazardous working conditions as mediating factors. SSM Popul. Health 2017, 3, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nature 2016, 538, 161–164. [Google Scholar] [CrossRef]
- Tournamille, C.; Colin, Y.; Cartron, J.P.; Van Kim, C.L. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy–negative individuals. Nat. Genet. 1995, 10, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.; Nalls, M.A.; Kao, W.H.L.; Akylbekova, E.L.; Tandon, A.; Patterson, N.; Mullikin, J.; Hsueh, W.C.; Cheng, C.Y.; Coresh, J.; et al. Reduced neutrophil count in people of African descent is due to a regulatory variant in the Duffy antigen receptor for chemokines gene. PLoS Genet. 2009, 5, e1000360. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Eliot, M.; Koestler, D.C.; Wu, W.C.; Kelsey, K.T. Association of neutrophil-to-lymphocyte ratio with mortality and cardiovascular disease in the jackson heart study and modification by the duffy antigen variant. JAMA Cardiol. 2018, 3, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ou, Z.L.; Hou, Y.F.; Luo, J.M.; Shen, Z.Z.; Ding, J.; Shao, Z.M. Enhanced expression of Duffy antigen receptor for chemokines by breast cancer cells attenuates growth and metastasis potential. Oncogene 2006, 25, 7201–7211. [Google Scholar] [CrossRef]
- Apostolakis, S.; Chalikias, G.K.; Tziakas, D.N.; Konstantinides, S. Erythrocyte Duffy antigen receptor for chemokines (DARC): Diagnostic and therapeutic implications in atherosclerotic cardiovascular disease. Acta Pharmacol. Sin. 2011, 32, 417–424. [Google Scholar] [CrossRef]
- Martini, R.; Chen, Y.; Jenkins, B.D.; Elhussin, I.A.; Cheng, E.; Hoda, S.A.; Ginter, P.S.; Hanover, J.; Zeidan, R.B.; Oppong, J.K.; et al. Investigation of triple-negative breast cancer risk alleles in an International African-enriched cohort. Sci. Rep. 2021, 11, 9247. [Google Scholar] [CrossRef]
- Charles, B.A.; Hsieh, M.M.; Adeyemo, A.A.; Shriner, D.; Ramos, E.; Chin, K.; Srivastava, K.; Zakai, N.A.; Cushman, M.; McClure, L.A.; et al. Analyses of genome wide association data, cytokines, and gene expression in African-Americans with benign ethnic neutropenia. PLoS ONE 2018, 13, e0194400. [Google Scholar] [CrossRef]
- Anderson, G.; Cummings, S.; Freedman, L.S.; Furberg, C.; Henderson, M.; Johnson, S.R.; Kuller, L.; Manson, J.; Oberman, A.; Prentice, R.L.; et al. Design of the Women’s Health Initiative clinical trial and observational study. Control Clin. Trials 1998, 19, 61–109. [Google Scholar]
- Carpenter, M.A.; Crow, R.; Steffes, M.; Rock, W.; Heilbraun, J.; Evans, G.; Skelton, T.; Jensen, R.; Sarpong, D. Laboratory, Reading Center, and Coordinating Center data management methods in the Jackson Heart Study. Am. J. Med. Sci. 2004, 328, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.A. The Jackson Heart Study: An overview. Ethn. Dis. 2005, 15, S6-1-3. [Google Scholar] [PubMed]
- Bild, D.E.; Bluemke, D.A.; Burke, G.L.; Detrano, R.; Diez Roux, A.V.; Folsom, A.R.; Greenland, P.; Jacobs, D.R.; Kronmal, R.; Liu, K.; et al. Multi-Ethnic Study of Atherosclerosis: Objectives and design. Am. J. Epidemiol. 2002, 156, 871–881. [Google Scholar] [CrossRef]
- Margolis, K.L.; Qi, L.; Brzyski, R.; Bonds, D.E.; Howard, B.V.; Kempainen, S.; Liu, S.; Robinson, J.G.; Safford, M.M.; Tinker, L.T.; et al. Validity of diabetes self-reports in the Women’s Health Initiative: Comparison with medication inventories and fasting glucose measurements. Clin. Trials 2008, 5, 240–247. [Google Scholar] [CrossRef]
- Fox, E.R.; Benjamin, E.J.; Sarpong, D.F.; Rotimi, C.N.; Wilson, J.G.; Steffes, M.W.; Chen, G.; Adeyemo, A.; Taylor, J.K.; Samdarshi, T.E.; et al. Epidemiology, Heritability, and Genetic Linkage of C-Reactive Protein in African Americans (from the Jackson Heart Study). Am. J. Cardiol. 2008, 102, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.D.; Cushman, M.; Nasir, K.; Bluemke, D.A.; Edvardsen, T.; Fernandes, V.; Lai, S.; Tracy, R.P.; Lima, J.A.C. Relationship Between C-Reactive Protein Levels and Regional Left Ventricular Function in Asymptomatic Individuals. The Multi-Ethnic Study of Atherosclerosis. J. Am. Coll. Cardiol. 2007, 49, 594–600. [Google Scholar] [CrossRef]
- Robins, M.; Weinfeld, F.D. The national survey of stroke. Study design and methodology. Stroke 1981, 12, I7–I11. [Google Scholar]
- Kawasaki, R.; Xie, J.; Cheung, N.; Lamoureux, E.; Klein, R.; Klein, B.E.K.; Cotch, M.F.; Sharrett, A.R.; Shea, S.; Wong, T.Y. Retinal microvascular signs and risk of stroke: The Multi-Ethnic Study of Atherosclerosis (MESA). Stroke 2012, 43, 3245–3251. [Google Scholar] [CrossRef]
- Curb, J.D.; Mctiernan, A.; Heckbert, S.R.; Kooperberg, C.; Stanford, J.; Nevitt, M.; Johnson, K.C.; Proulx-Burns, L.; Pastore, L.; Criqui, M.; et al. Outcomes ascertainment and adjudication methods in the women’s health initiative. Ann. Epidemiol. 2003, 13, S122–S128. [Google Scholar] [CrossRef]
- Rosamond, W.D.; Chang, P.P.; Baggett, C.; Johnson, A.; Bertoni, A.G.; Shahar, E.; Deswal, A.; Heiss, G.; Chambless, L.E. Classification of heart failure in the atherosclerosis risk in communities (ARIC) study a comparison of diagnostic criteria. Circ. Heart Fail. 2012, 5, 152–159. [Google Scholar] [CrossRef]
- Raffield, L.M.; Iyengar, A.K.; Wang, B.; Gaynor, S.M.; Spracklen, C.N.; Zhong, X.; Kowalski, M.H.; Salimi, S.; Polfus, L.M.; Benjamin, E.J.; et al. Allelic Heterogeneity at the CRP Locus Identified by Whole-Genome Sequencing in Multi-ancestry Cohorts. Am. J. Hum. Genet. 2020, 106, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.; McGuire, D.K.; Murphy, S.A.; Stanek, H.G.; Das, S.R.; Vongpatanasin, W.; Wians, F.H.; Grundy, S.M.; De Lemos, J.A. Race and gender differences in C-reactive protein levels. J. Am. Coll. Cardiol. 2005, 46, 464–469. [Google Scholar] [CrossRef] [PubMed]
- King, D.E.; Mainous, A.G.; Buchanan, T.A.; Pearson, W.S. C-reactive protein and glycemic control in adults with diabetes. Diabetes Care 2003, 26, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.; Bouter, L.M.; McQuillan, G.M.; Wener, M.H.; Harris, T.B. Elevated C-reactive protein levels in overweight and obese adults. J. Am. Med. Assoc. 1999, 282, 2131–2135. [Google Scholar] [CrossRef]
- Farmer, H.R.; Wray, L.A.; Haas, S.A. Race, Gender, and Socioeconomic Variations in C-Reactive Protein Using the Health and Retirement Study. J. Gerontol.-Ser. B Psychol. Sci. Soc. Sci. 2021, 76, 583–595. [Google Scholar] [CrossRef]
- Lange, L.A.; Carlson, C.S.; Hindorff, L.A.; Lange, E.M.; Walston, J.; Durda, J.P.; Cushman, M.; Bis, J.C.; Zeng, D.; Lin, D.; et al. Association of polymorphisms in the CRP gene with circulating C-reactive protein levels and cardiovascular events. JAMA 2006, 296, 2703–2711. [Google Scholar] [CrossRef]
- Grann, V.R.; Ziv, E.; Joseph, C.K.; Neugut, A.I.; Wei, Y.; Jacobson, J.S.; Horwitz, M.S.; Bowman, N.; Beckmann, K.; Hershman, D.L. Duffy (Fy), DARC, and neutropenia among women from the United States, Europe and the Caribbean. Br. J. Haematol. 2008, 143, 288–293. [Google Scholar] [CrossRef]
- Reiner, A.P.; Beleza, S.; Franceschini, N.; Auer, P.L.; Robinson, J.G.; Kooperberg, C.; Peters, U.; Tang, H. Genome-wide association and population genetic analysis of c-reactive protein in african american and hispanic american women. Am. J. Hum. Genet. 2012, 91, 502–512. [Google Scholar] [CrossRef]
- Jinna, N.; Rida, P.; Su, T.; Gong, Z.; Yao, S.; LaBarge, M.; Natarajan, R.; Jovanovic-Talisman, T.; Ambrosone, C.; Seewaldt, V. The DARC Side of Inflamm-Aging: Duffy Antigen Receptor for Chemokines (DARC/ACKR1) as a Potential Biomarker of Aging, Immunosenescence, and Breast Oncogenesis among High-Risk Subpopulations. Cells 2022, 11, 3818. [Google Scholar] [CrossRef]
- Ligthart, S.; Vaez, A.; Võsa, U.; Stathopoulou, M.G.; de Vries, P.S.; Prins, B.P.; Van der Most, P.J.; Tanaka, T.; Naderi, E.; Rose, L.M.; et al. Genome Analyses of >200,000 Individuals Identify 58 Loci for Chronic Inflammation and Highlight Pathways that Link Inflammation and Complex Disorders. Am. J. Hum. Genet. 2018, 103, 691–706. [Google Scholar] [CrossRef]
- Song, M.; Graubard, B.I.; Rabkin, C.S.; Engels, E.A. Neutrophil-to-lymphocyte ratio and mortality in the United States general population. Sci. Rep. 2021, 11, 464. [Google Scholar] [CrossRef] [PubMed]
- Bihlmeyer, N.A.; Brody, J.A.; Smith, A.V.; Lunetta, K.L.; Nalls, M.; Smith, J.A.; Tanaka, T.; Davies, G.; Yu, L.; Mirza, S.S.; et al. Genetic diversity is a predictor of mortality in humans. BMC Genet. 2014, 15, 159. [Google Scholar] [CrossRef] [PubMed]
- Palmblad, J.; Sohlberg, E.; Nilsson, C.C.; Lindqvist, H.; Deneberg, S.; Ratcliffe, P.; Meinke, S.; Mörtberg, A.; Klimkowska, M.; Höglund, P. Clinical and immunological features in ACKR1/DARC-associated neutropenia. Blood Adv. 2024, 8, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Hershman, D.; McBride, R.; Jacobson, J.S.; Lamerato, L.; Roberts, K.; Grann, V.R.; Neugut, A.I. Racial disparities in treatment and survival among women with early-stage breast cancer. J. Clin. Oncol. 2005, 23, 6639–6646. [Google Scholar] [CrossRef]
- DeNavas-Walt, C.; Cleveland, R.W.; Roemer, M.I. Money Income in the United States: 2000; United States Census Bureau: Suitland-Silver HillSuitland, MD, USA, 2001. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WHI | |||
|---|---|---|---|
| Duffy-antigen-receptor-negative (n = 5959) | Duffy-antigen-receptor-positive (n = 3891) | p-value | |
| Demographics | |||
| Age (years) | 61.2 ± 7.0 | 62.0 ± 7.2 | <0.001 |
| Males (%) | - | - | - |
| Income tier | 2.0 ± 0.8 | 2.1 ± 0.8 | <0.001 |
| Education tier | 1.6 ± 0.7 | 1.7 ± 0.6 | <0.001 |
| Clinical variables | |||
| Systolic blood pressure | 132.3 ± 17.8 | 131.7 ± 17.8 | 0.31 |
| Diabetes | 917 (15) | 531 (14) | 0.02 |
| Current smoker | 661 (11) | 429 (11) | 0.97 |
| BMI (kg/m2) | 31.2 ± 6.7 | 31.1 ± 6.7 | 0.54 |
| hs-CRP (mg/L) | 6.6 ± 7.9 | 5.6 ± 8.3 | <0.001 |
| JHS | |||
| Duffy-antigen-receptor-negative (n = 2389) | Duffy-antigen-receptor-positive (n = 1017) | p-value | |
| Demographics | |||
| Age (years) | 54.7 ± 12.8 | 56.0 ± 12.8 | 0.04 |
| Males (%) | 890 (37) | 374 (37) | 0.79 |
| Income tier | 2.7 ± 1.0 | 2.8 ± 1.0 | 0.10 |
| Education tier | 1.4 ± 0.8 | 1.5 ± 0.7 | 0.008 |
| Clinical variables | |||
| Systolic blood pressure | 127.4 ± 17.8 | 126.5 ± 16.5 | 0.18 |
| Diabetes | 596 (25) | 235 (23) | 0.25 |
| Current smoker | 318 (13) | 127 (12) | 0.51 |
| BMI (kg/m2) | 31.9 ± 7.2 | 31.7 ± 7.5 | 0.72 |
| hs-CRP (mg/L) | 5.6 ± 10.8 | 4.4 ± 6.9 | <0.001 |
| MESA | |||
| Duffy-antigen-receptor-negative (n = 675) | Duffy-antigen-receptor-positive (n = 427) | p-value | |
| Demographics | |||
| Age (years) | 60.6 ± 9.5 | 61.4 ± 9.7 | 0.49 |
| Males (%) | 317 (47) | 201 (47) | 0.97 |
| Income tier | 2.3 ± 0.9 | 2.4 ± 0.9 | 0.07 |
| Education tier | 2.6 ± 0.7 | 2.6 ± 0.6 | 0.10 |
| Clinical variables | |||
| Systolic blood pressure | 130.4 ± 21.1 | 128.9 ± 19.5 | 0.74 |
| Diabetes | 101 (15) | 75 (18) | 0.24 |
| Current smoker | 124 (18) | 74 (17) | 0.66 |
| BMI (kg/m2) | 29.9 ± 5.4 | 30.0 ± 5.4 | 0.24 |
| hs-CRP (mg/L) | 4.0 ± 5.4 | 3.6 ± 6.2 | 0.02 |
| (a) | |||||
| Estimate (B) | Std. Error | p-Value | |||
| WHI (n = 6102) | Model 1 * | 0.189 | 0.031 | 1.09 × 10−9 | |
| Model 2 ** | 0.061 | 0.035 | 0.08 | ||
| JHS (n = 2863) | Model 1 | 0.293 | 0.049 | 3.38 × 10−9 | |
| Model 2 | 0.129 | 0.057 | 0.02 | ||
| MESA (n = 830) | Model 1 | 0.201 | 0.085 | 0.02 | |
| Model 2 | 0.092 | 0.098 | 0.35 | ||
| Meta-analysis | Model 1 | 0.18 | 0.069 | 2.62 × 10−9 | |
| Model 2 | 0.08 | 0.03 | 0.005 | ||
| (b) | |||||
| Estimate (B) | Standard Error | Hazard Ratio | p-Value | ||
| WHI (n = 10,083) | Model 1 * | 0.140 | 0.059 | 1.50 [1.03, 1.29] | 0.02 |
| Model 2 ** | 0.085 | 0.063 | 1.09 [0.96, 1.23] | 0.18 | |
| JHS (n = 3406) | Model 1 | 0.101 | 0.088 | 1.11 [0.93, 1.31] | 0.25 |
| Model 2 | 0.035 | 0.099 | 1.04 [0.85, 1.25] | 0.72 | |
| MESA (n = 1102) | Model 1 | −0.195 | 0.167 | 0.82 [0.59, 1.14] | 0.24 |
| Model 2 | −0.246 | 0.175 | 0.78 [0.55, 1.10] | 0.16 | |
| Hazard Ratio [95% CI] | p-Value | |
|---|---|---|
| Model 1 (n = 8821, n = 529 events) | 0.81 [0.61, 1.06] | 0.12 |
| Model 2 (n = 6973, 358 events) | 0.75 [0.54, 1.05] | 0.09 |
| Model 3 (n = 6527, 340 events) | 0.73 [0.53, 1.01] | 0.06 |
| (a) | ||||||||||
| Coronary Heart Disease (1025 Events, 6592 Censored) | Stroke (331 Events, 7282 Censored) | Infectious Disease (221 Events, 7359 Censored) | Cancer (965 Events, 6646 Censored) | Alzheimer/Dementia (349 Events, 6812 Censored) | ||||||
| Hazard Ratio [95% CI] | p-value | Hazard Ratio [95% CI] | p-value | Hazard Ratio [95% CI] | p-value | Hazard Ratio [95% CI] | p-value | Hazard Ratio [95% CI] | p-value | |
| Model 1 | 1.20 [1.05, 1.38] | 0.007 | 1.11 [0.88, 1.40] | 0.38 | 1.15 [0.87, 1.53] | 0.33 | 1.01 [0.88, 1.15] | 0.89 | 0.97 [0.78, 1.21] | 0.81 |
| Model 2 | 1.11 [0.98, 1.28] | 0.11 | 1.05 [0.84, 1.33] | 0.66 | 1.06 [0.80, 1.42] | 0.66 | 0.99 [0.86, 1.14] | 0.94 | 0.94 [0.76, 1.18] | 0.64 |
| Model 3 | 1.09 [0.96, 1.25] | 0.17 | 1.05 [0.83, 1.33] | 0.68 | 1.04 [0.78, 1.39] | 0.76 | 0.99 [0.86, 1.13] | 0.89 | 0.96 [0.76, 1.20] | 0.72 |
| (b) | ||||||||||
| Coronary Heart Disease (54 Events, 975 Censored) | ||||||||||
| Hazard Ratio [95% CI] | p-value | |||||||||
| Model 1 | 2.11 [1.14, 3.92] | 0.02 | ||||||||
| Model 2 | 2.04 [1.08, 3.80] | 0.03 | ||||||||
| Model 3 | 2.14 [1.13, 4.04] | 0.02 | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, E.T.; Haessler, J.; Taylor, K.D.; Tuftin, B.; Briggs, M.; Parikh, M.A.; Peterson, S.J.; Gerszten, R.E.; Wilson, J.G.; Kelsey, K.; et al. The Relationship of Duffy Gene Polymorphism with High-Sensitivity C-Reactive Protein, Mortality, and Cardiovascular Outcomes in Black Individuals. Genes 2024, 15, 1382. https://doi.org/10.3390/genes15111382
Ha ET, Haessler J, Taylor KD, Tuftin B, Briggs M, Parikh MA, Peterson SJ, Gerszten RE, Wilson JG, Kelsey K, et al. The Relationship of Duffy Gene Polymorphism with High-Sensitivity C-Reactive Protein, Mortality, and Cardiovascular Outcomes in Black Individuals. Genes. 2024; 15(11):1382. https://doi.org/10.3390/genes15111382
Chicago/Turabian StyleHa, Edward T., Jeffery Haessler, Kent D. Taylor, Bjoernar Tuftin, Matt Briggs, Manish A. Parikh, Stephen J. Peterson, Robert E. Gerszten, James G. Wilson, Karl Kelsey, and et al. 2024. "The Relationship of Duffy Gene Polymorphism with High-Sensitivity C-Reactive Protein, Mortality, and Cardiovascular Outcomes in Black Individuals" Genes 15, no. 11: 1382. https://doi.org/10.3390/genes15111382
APA StyleHa, E. T., Haessler, J., Taylor, K. D., Tuftin, B., Briggs, M., Parikh, M. A., Peterson, S. J., Gerszten, R. E., Wilson, J. G., Kelsey, K., Tahir, U. A., Seeman, T., Rich, S. S., Carson, A. P., Post, W. S., Kooperberg, C., Rotter, J. I., Raffield, L. M., Auer, P., & Reiner, A. P. (2024). The Relationship of Duffy Gene Polymorphism with High-Sensitivity C-Reactive Protein, Mortality, and Cardiovascular Outcomes in Black Individuals. Genes, 15(11), 1382. https://doi.org/10.3390/genes15111382