
Gene Expression Profiling and Immune Pathway Dysregulation in Ribonucleoprotein Autoantibody-Positive Systemic Lupus Erythematosus Patients

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Data Processing
2.3. Functional Enrichment Analysis
2.4. PPI Network Building
2.5. Tissue Enrichment Analysis
2.6. Gene Frequency Analysis
2.7. Cell Type Annotation
3. Results
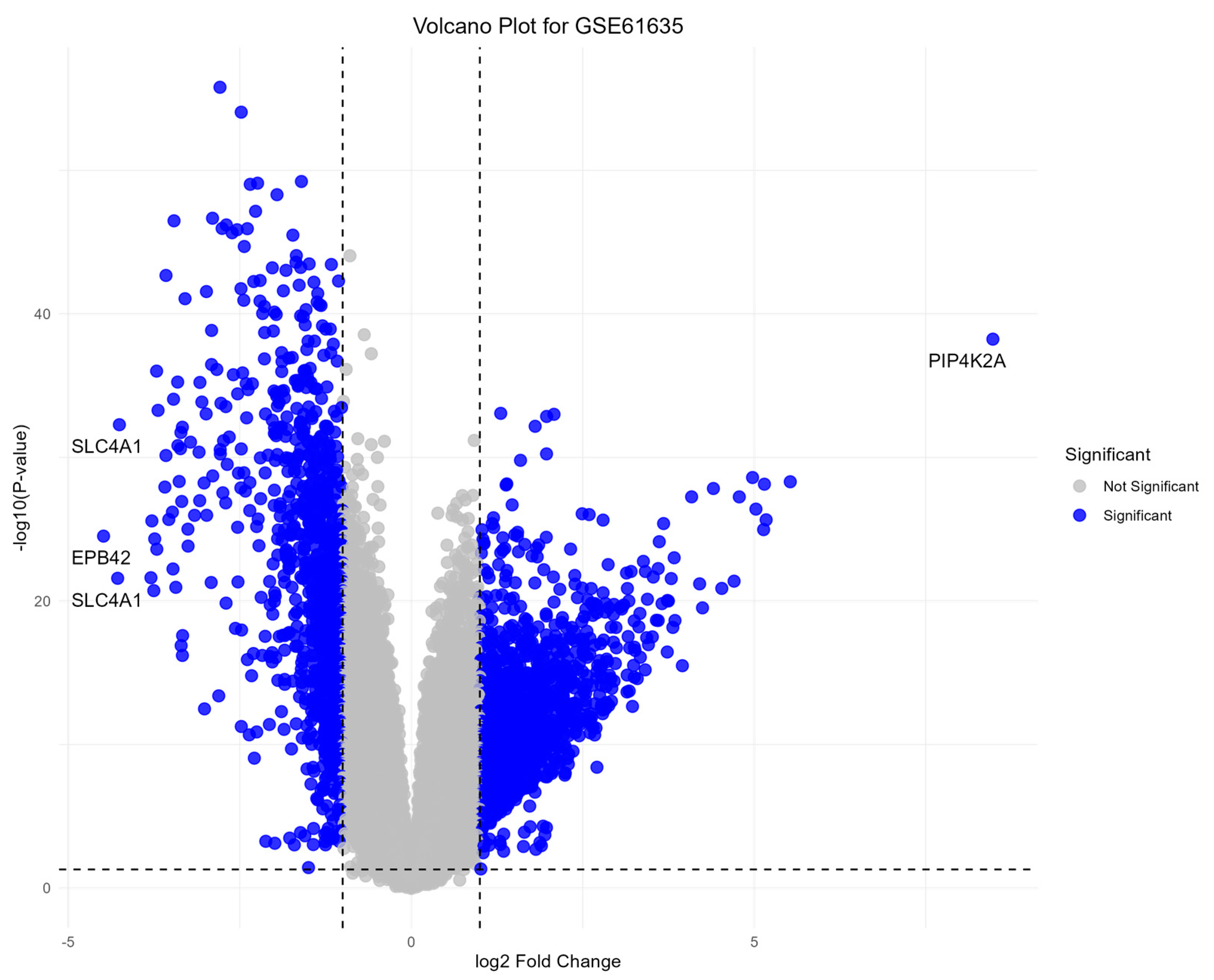
3.1. Differential Gene Expression Analysis
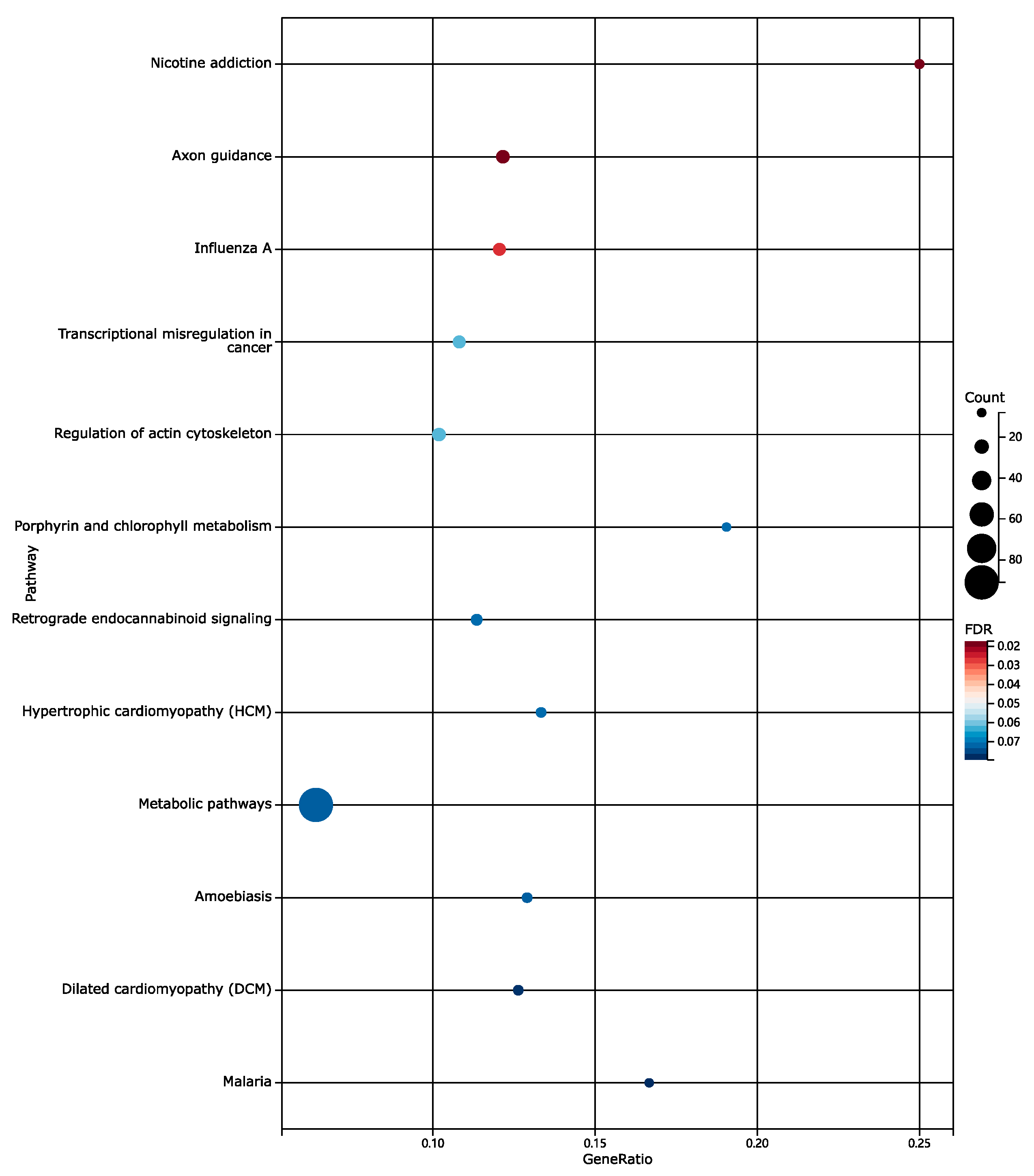
3.2. Functional Enrichment Analysis Results
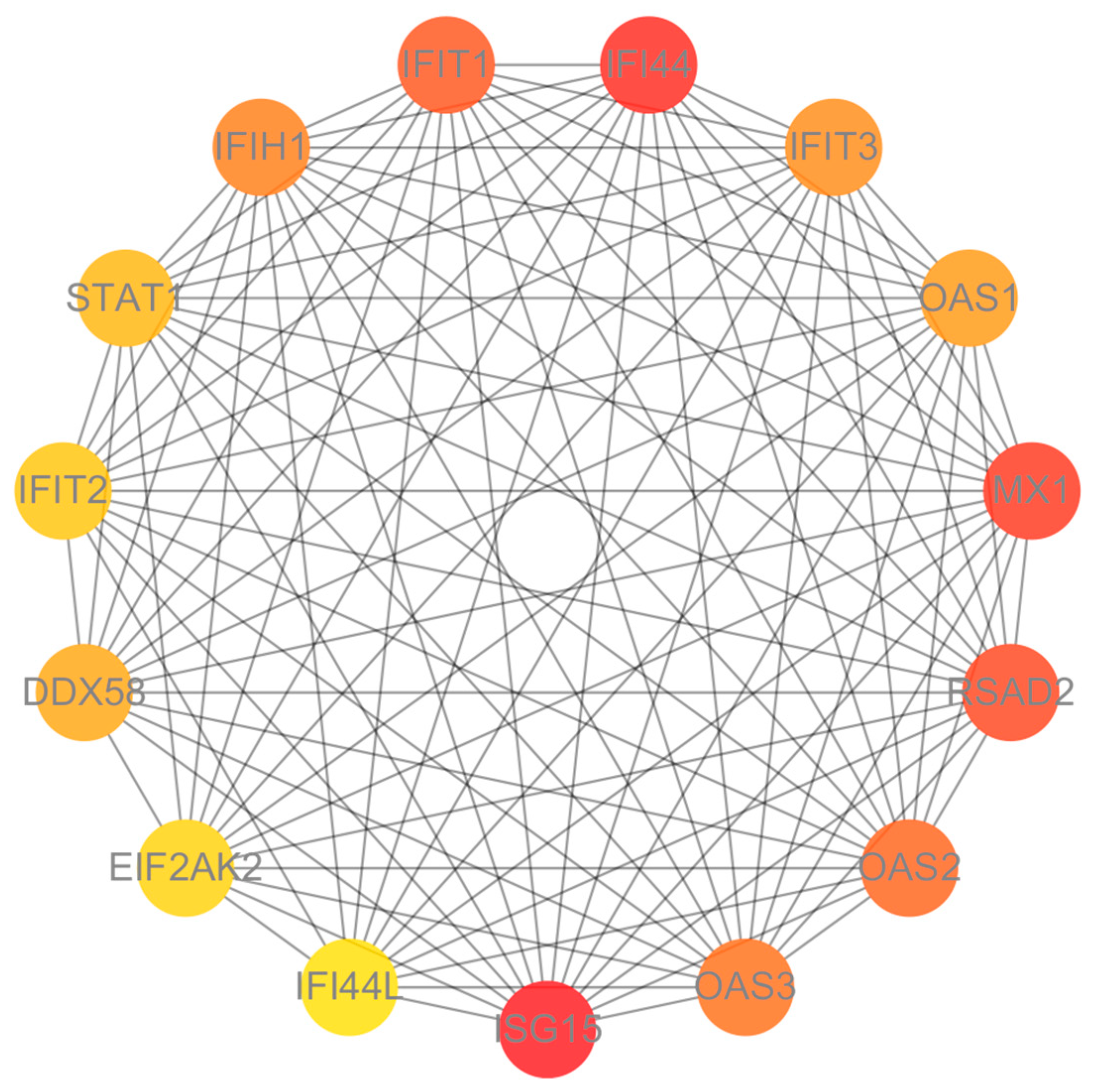
3.3. Protein–Protein Interaction (PPI) Network Analysis
3.4. Tissue Enrichment Analysis Results
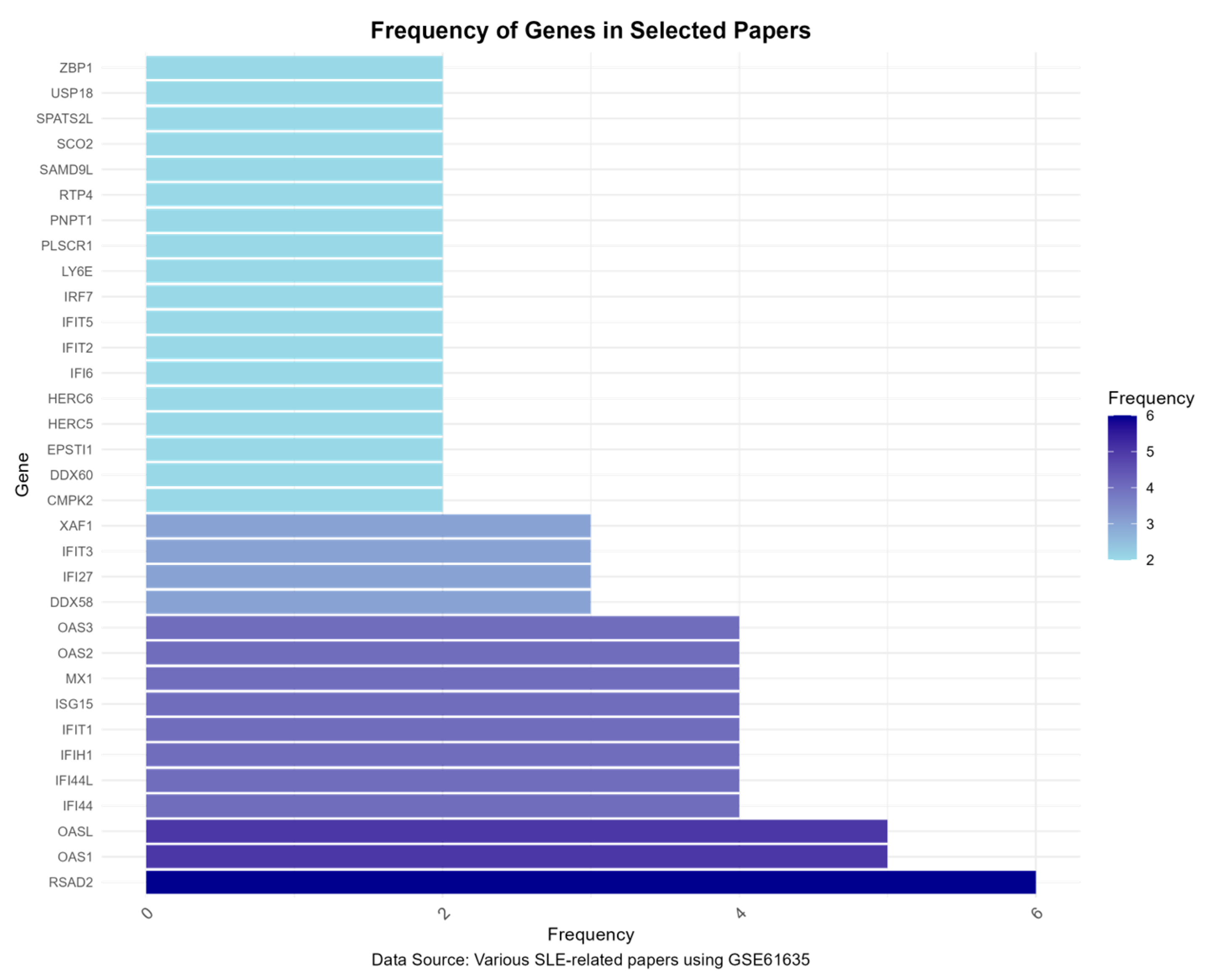
3.5. Gene Frequency Analysis Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ameer, M.A.; Chaudhry, H.; Mushtaq, J.; Khan, O.S.; Babar, M.; Hashim, T.; Zeb, S.; Tariq, M.A.; Patlolla, S.R.; Ali, J.; et al. An Overview of Systemic Lupus Erythematosus (SLE) Pathogenesis, Classification, and Management. Cureus 2022, 14, e30330. [Google Scholar] [CrossRef]
- Antibodies to Double-Stranded (ds)DNA, sm, and U1 RNP—UpToDate. Available online: https://www.uptodate.com/contents/antibodies-to-double-stranded-ds-dna-sm-and-u1-rnp#H158502 (accessed on 13 October 2024).
- Fu, Q.; Zhang, X. From blood to tissue: Take a deeper look at B cells in lupus. Cell. Mol. Immunol. 2021, 18, 2073–2074. [Google Scholar] [CrossRef]
- Suárez-Fueyo, A.; Bradley, S.J.; Tsokos, G.C. T cells in Systemic Lupus Erythematosus. Curr. Opin. Immunol. 2016, 43, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Pisetsky, D.S. Evolving Story of Autoantibodies in Systemic Lupus Erythematosus. J. Autoimmun. 2020, 110, 102356. [Google Scholar] [CrossRef] [PubMed]
- Rönnblom, L.; Pascual, V. The innate immune system in SLE: Type I interferons and dendritic cells. Lupus 2008, 17, 394–399. [Google Scholar] [CrossRef]
- Pathak, S.; Mohan, C. Cellular and molecular pathogenesis of systemic lupus erythematosus: Lessons from animal models. Arthritis Res. Ther. 2011, 13, 241. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhan, F.; Lin, S. Clinical Values of the Identified Hub Genes in Systemic Lupus Erythematosus. Front. Immunol. 2022, 13, 844025. [Google Scholar] [CrossRef]
- Suszek, D.; Dubaj, M.; Bigosiński, K.; Dembowska, A.; Kaniewski, M.; Sielwanowska, W.; Skierkowski, B.; Dzikowska, I.; Sieczka, J.; Majdan, M. Usefulness in daily practice of the systemic lupus erythematosus disease activity index 2000 scale and the systemic lupus erythematosus disease activity score index for assessing the activity of systemic lupus erythematosus. Reumatologia 2024, 62, 187. [Google Scholar] [CrossRef]
- Jain, A.; Tuteja, G. TissueEnrich: Tissue-specific gene enrichment analysis. Bioinformatics 2019, 35, 1966–1967. [Google Scholar] [CrossRef]
- Chen, H.; Huang, L.; Jiang, X.; Wang, Y.; Bian, Y.; Ma, S.; Liu, X. Establishment and analysis of a disease risk prediction model for the systemic lupus erythematosus with random forest. Front. Immunol. 2022, 13, 1025688. [Google Scholar] [CrossRef]
- Li, H.; Zhou, L.; Zhou, W.; Zhang, X.; Shang, J.; Feng, X.; Yu, L.; Fan, J.; Ren, J.; Zhang, R.; et al. Decoding the mitochondrial connection: Development and validation of biomarkers for classifying and treating systemic lupus erythematosus through bioinformatics and machine learning. BMC Rheumatol. 2023, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Guan, X.; Wang, Y.; Lv, Y.; Wu, Y.; Yu, M.; Sun, Y. Cuproptosis-related gene identification and immune infiltration analysis in systemic lupus erythematosus. Front. Immunol. 2023, 14, 1157196. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yang, J.; He, Q.; Zhang, Z.; Xu, G. Comprehensive bioinformatics analysis reveals the crosstalk genes and immune relationship between the systemic lupus erythematosus and venous thromboembolism. Front. Immunol. 2023, 14, 1196064. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, L.; Hu, L.-L.; Yu, L.-L.; Li, J.-P.; Rao, J.; Zhu, L.-J.; Liang, Q.; Zhang, R.-W.; Bao, H.-H.; et al. Potential therapeutic target genes for systemic lupus erythematosus: A bioinformatics analysis. Bioengineered 2021, 12, 2810–2819. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Tan, L.; Zhang, F.; Li, S.; Lai, Z.; Xu, H.; Xiong, Z.; Dai, Y. The circRNA–miRNA–mRNA regulatory network in plasma and peripheral blood mononuclear cells and the potential associations with the pathogenesis of systemic lupus erythematosus. Clin. Rheumatol. 2023, 42, 1885–1896. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhang, W.; Hong, X.; Zeng, Z.; Chen, Y.; Liao, S.; Cai, W.; Xu, Y.; Wang, G.; Liu, D.; et al. Screening Biomarkers for Systemic Lupus Erythematosus Based on Machine Learning and Exploring Their Expression Correlations with the Ratios of Various Immune Cells. Front. Immunol. 2022, 13, 873787. [Google Scholar] [CrossRef]
- Zhu, Q.-Y. Bioinformatics analysis of the pathogenic link between Epstein-Barr virus infection, systemic lupus erythematosus and diffuse large B cell lymphoma. Sci. Rep. 2023, 13, 6310. [Google Scholar] [CrossRef]
- How Does Lupus Affect Fertility? Verywell Health. Available online: https://www.verywellhealth.com/lupus-and-infertility-5114589 (accessed on 1 October 2024).
- How Lupus Affects the Nervous System|Lupus Foundation of America. Available online: https://www.lupus.org/resources/how-lupus-affects-the-nervous-system (accessed on 1 October 2024).
- NCBI Gene. SLC4A1: Solute Carrier Family 4 Member 1 (Diego Blood Group); National Center for Biotechnology Information (NCBI): Bethesda, MD, USA, 2008. Available online: https://www.ncbi.nlm.nih.gov/gene/6521 (accessed on 1 October 2024).
- NCBI Gene. EPB42: Erythrocyte Membrane Protein Band 4.2; National Center for Biotechnology Information (NCBI): Bethesda, MD, USA, 2008. Available online: https://www.ncbi.nlm.nih.gov/gene/2038 (accessed on 1 October 2024).
- Herrada, A.A.; Escobedo, N.; Iruretagoyena, M.; Valenzuela, R.A.; Burgos, P.I.; Cuitino, L.; Llanos, C. Innate Immune Cells’ Contribution to Systemic Lupus Erythematosus. Front. Immunol. 2019, 10, 772. [Google Scholar] [CrossRef]
- Kaplan, M.J. Role of neutrophils in systemic autoimmune diseases. Arthritis Res. Ther. 2013, 15, 219. [Google Scholar] [CrossRef]
- Wirestam, L.; Arve, S.; Linge, P.; Bengtsson, A.A. Neutrophils—Important Communicators in Systemic Lupus Erythematosus and Antiphospholipid Syndrome. Front. Immunol. 2019, 10, 2734. [Google Scholar] [CrossRef]
- Gupta, S.; Kaplan, M.J. Bite of the wolf: Innate immune responses propagate autoimmunity in lupus. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, S.; Damodaran, T. Gene Expression Profiling and Immune Pathway Dysregulation in Ribonucleoprotein Autoantibody-Positive Systemic Lupus Erythematosus Patients. Genes 2024, 15, 1353. https://doi.org/10.3390/genes15111353
Tan S, Damodaran T. Gene Expression Profiling and Immune Pathway Dysregulation in Ribonucleoprotein Autoantibody-Positive Systemic Lupus Erythematosus Patients. Genes. 2024; 15(11):1353. https://doi.org/10.3390/genes15111353
Chicago/Turabian StyleTan, Siyuan, and Tirupapuliyur Damodaran. 2024. "Gene Expression Profiling and Immune Pathway Dysregulation in Ribonucleoprotein Autoantibody-Positive Systemic Lupus Erythematosus Patients" Genes 15, no. 11: 1353. https://doi.org/10.3390/genes15111353
APA StyleTan, S., & Damodaran, T. (2024). Gene Expression Profiling and Immune Pathway Dysregulation in Ribonucleoprotein Autoantibody-Positive Systemic Lupus Erythematosus Patients. Genes, 15(11), 1353. https://doi.org/10.3390/genes15111353