

Comparative Transcriptome Analysis of High- and Low-Growth Genotypes of Eucalyptus urophylla in Response to Long-Term Nitrogen Deficiency
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Nutrient Treatments
2.2. Determination of Growth Characteristics
2.3. RNA Isolation, Sequencing, and Assembly
2.4. Normalization of Gene Expression Levels and Identification of DEGs
2.4.1. Functional Annotation and GO and KEGG Classification
2.4.2. Validation of the Expression Level
3. Results
3.1. Effect of Different Nutrient Treatments on Tree Growth Characteristics
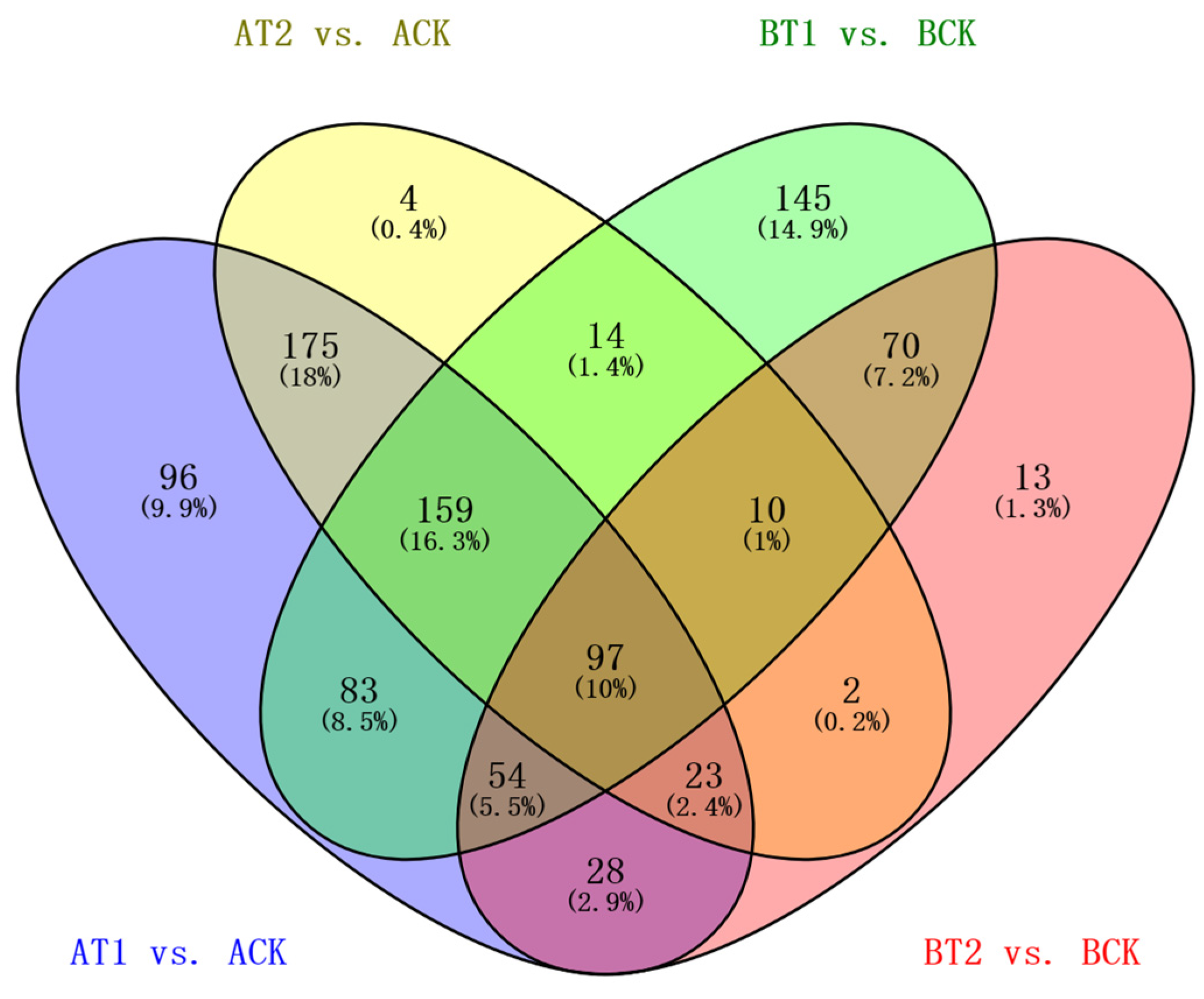
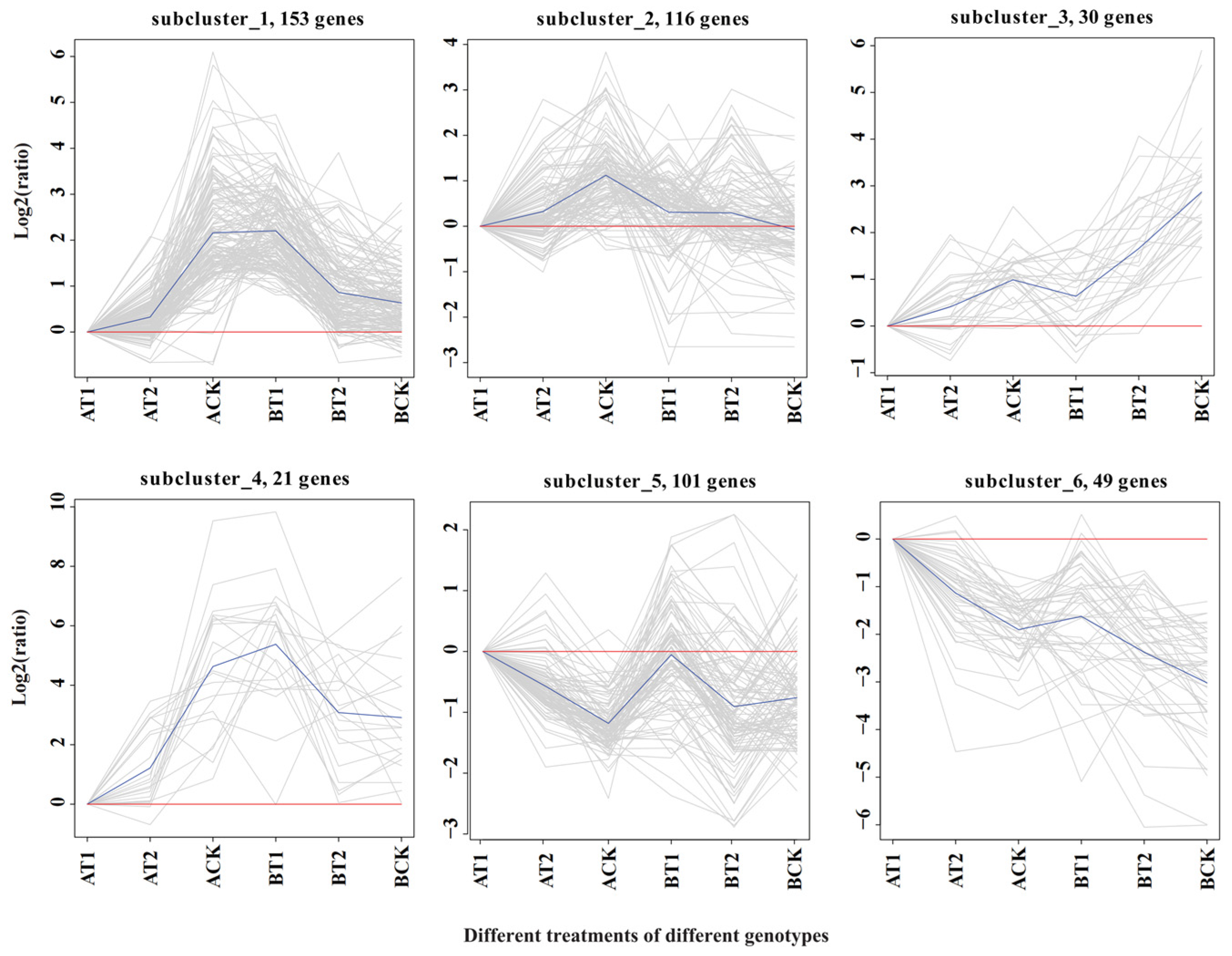
3.2. An RNA Sequencing Approach for the Assembly, Quantification, Identification, and Clustering of DEGs in Response to Nutrient Treatments
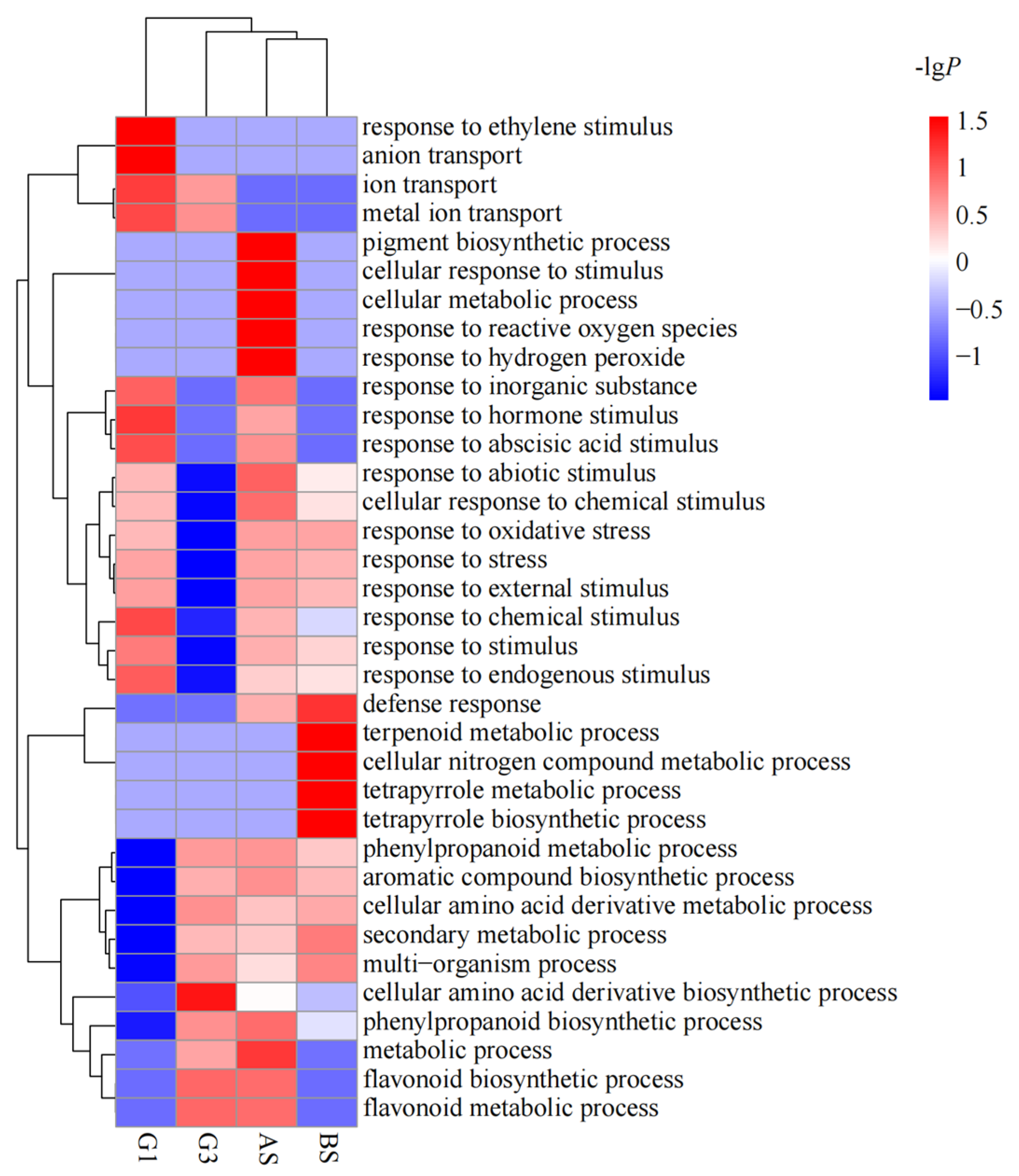
3.3. Functional Enrichment of the DEGs of Different Genotypes in Response to Nutrient Treatments
3.4. DEGs Involved in Plant Hormone Signal Transduction
3.5. Transcription Factors (TFs) Responding to Nutrient Deficiency
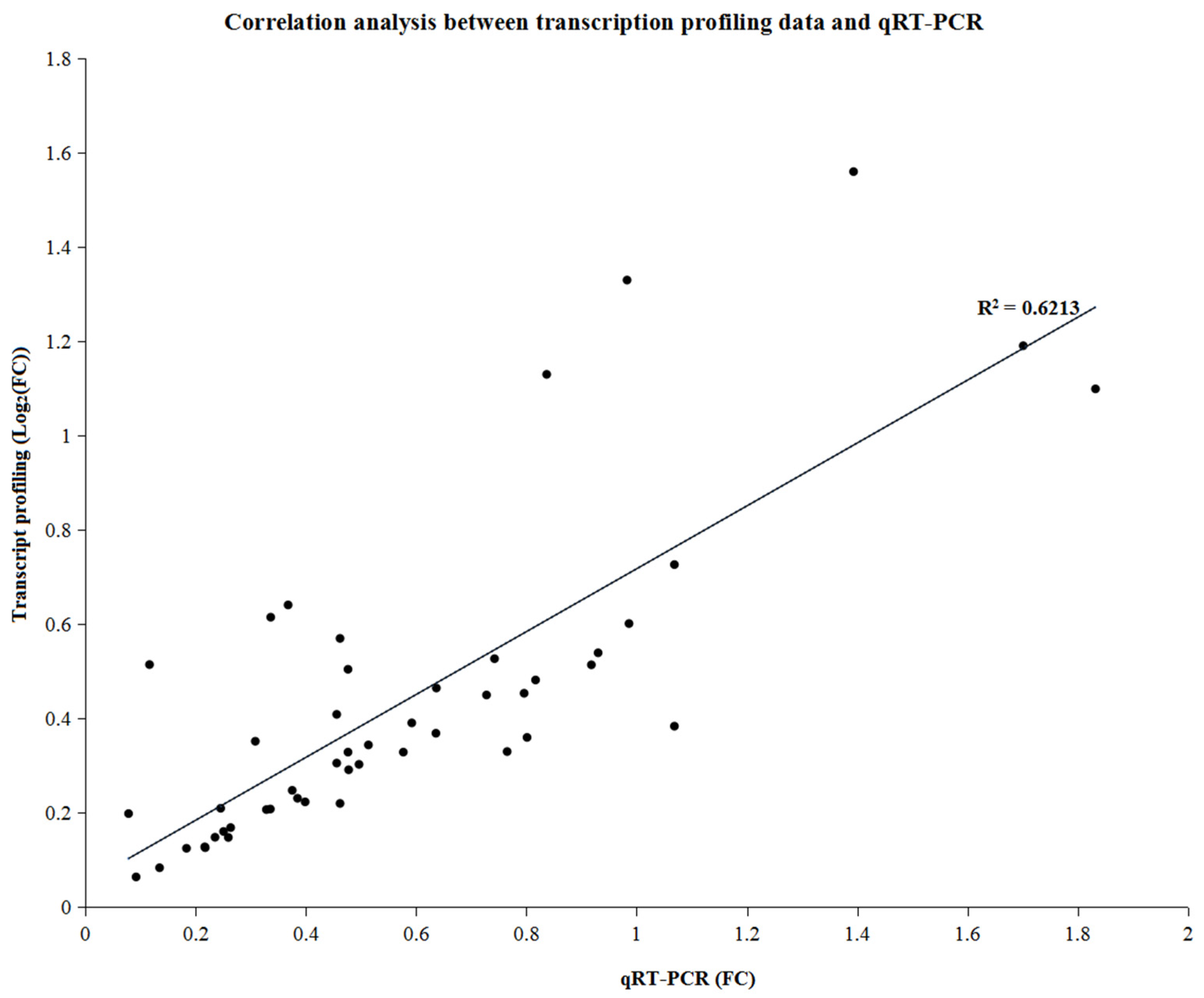
3.6. Validation of RNA Sequencing Results via Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maathuis, F.J. Physiological functions of mineral macronutrients. Curr. Opin. Plant Biol. 2009, 12, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Brix, H. Effects of thinning and nitrogen fertilization on growth of Douglas-fir: Relative contribution of foliage quantity and efficiency. Can. J. For. Res. 1983, 13, 167–175. [Google Scholar] [CrossRef]
- Gerloff, G.C. Comparative Mineral Nutrition of Plants. Annu. Rev. Plant Physiol. 1963, 14, 107–124. [Google Scholar] [CrossRef]
- Siddiqi, M.Y.; Glass, A.D.M. Utilization index: A modified approach to the estimation and comparison of nutrient utilization efficiency in plants. J. Plant Nutr. 2008, 4, 289–302. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Armengaud, P.; Breitling, R.; Amtmann, A. The Potassium-Dependent Transcriptome of Arabidopsis Reveals a Prominent Role of Jasmonic Acid in Nutrient Signaling. Plant Physiol. 2004, 136, 2556–2576. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Fang, Y.; Xiao, Y.; Jin, Y.; Ma, X.; Zhao, Y.; He, K.; Zhao, H.; Wang, H. Comparative transcriptome analysis to investigate the high starch accumulation of duckweed (Landoltia punctata) under nutrient starvation. Biotechnol. Biofuels 2013, 6, 72. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, S.; Yuan, M.; Zhou, K. De novo transcriptome assembly for pericarp in Litchi chinesis Sonn. cv. Feizixiao and identification of differentially expressed genes in response to Mg Foliar Nutrient. Sci. Hortic. 2017, 226, 59–67. [Google Scholar] [CrossRef]
- Pandey, G.K.; Cheong, Y.H.; Kim, K.; Grant, J.J.; Li, L.; Hung, W.; D’Angelo, C.; Weinl, S.; Kudla, J.; Luan, S. The Calcium Sensor Calcineurin B-Like 9 Modulates Abscisic Acid Sensitivity and Biosynthesis in Arabidopsis. Plant Cell 2004, 16, 1912–1924. [Google Scholar] [CrossRef]
- Poorter, L.; Markesteijn, L. Seedling Traits Determine Drought Tolerance of Tropical Tree Species. Biotropica 2008, 40, 321–331. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Garber, M.; Levin, J.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.; Gnirke, A.; Nusbaum, C.; et al. Correction: Corrigendum: Ab initio reconstruction of cell type–specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 756. [Google Scholar]
- Trapnell, C.; Williams, B.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.; Salzberg, S.; Wold, B.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Dal Santo, S.; Palliotti, A.; Zenoni, S.; Tornielli, G.B.; Fasoli, M.; Paci, P.; Tombesi, S.; Frioni, T.; Silvestroni, O.; Bellincontro, A.; et al. Distinct transcriptome responses to water limitation in isohydric and anisohydric grapevine cultivars. BMC Genom. 2016, 17, 815. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.; Lin, T.; Qin, M.; Peng, M.; Yang, C.; et al. Rewiring of the Fruit Metabolome in Tomato Breeding. Cell 2018, 172, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Connorton, J.; Balk, J. Iron Biofortification of Staple Crops: Lessons and Challenges in Plant Genetics. Plant Cell Physiol. 2019, 60, 1447–1456. [Google Scholar] [CrossRef]
- Li, D.; Wang, H.; Wang, M.; Li, G.; Chen, Z.; Leiser, W.L.; Wei, T.M.; Lu, X.; Wang, M.; Chen, S.; et al. Genetic Dissection of Phosphorus Use Efficiency in a Maize Association Population under Two P Levels in the Field. Int. J. Mol. Sci. 2021, 22, 9311. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, M.; Niu, X.; Wang, S.; Xu, Q.; Feng, Y.; Wang, C.; Deng, H.; Yuan, X.; Yu, H.; et al. Genetic variation and association mapping for 12 agronomic traits in indica rice. BMC Genom. 2015, 16, 1067. [Google Scholar] [CrossRef]
- Kothari, D.; Pargaien, N.; Tewari, L.M.; Dikshit, H.; Mishra, G.; Aski, M.; Bansal, R.; Gupta, S.; Kumar, S.; Nair, R. Genetic Variation for Traits Related to Phosphorus Use Efficiency in Vigna Species. Agronomy 2023, 13, 305. [Google Scholar] [CrossRef]
- Zulfiqar, A.; Naseer, S.; Saleem, A.; Sabar, M.; Ahmed, S.; Sardar, R.; Shahzadi, F.; Raza, Q. Genetic diversity studies for grain iron and zinc content analysis for Elite rice (Oryza sativa L.) genotype by using SSR markers. J. Food Compos. Anal. 2023, 115, 104816. [Google Scholar] [CrossRef]
- Niu, Y.; Li, J.; Sun, F.; Song, T.; Han, B.; Liu, Z.; Su, P. Comparative transcriptome analysis reveals the key genes and pathways involved in drought stress response of two wheat (Triticum aestivum L.) varieties. Genomics 2023, 115, 110688. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Jha, S.K.; Bagri, J.; Pandey, G.K. ABA Inducible Rice Protein Phosphatase 2C Confers ABA Insensitivity and Abiotic Stress Tolerance in Arabidopsis. PLoS ONE 2015, 10, e125168. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G. The environmental contribution to gene expression profiles. Nat. Rev. Genet. 2008, 9, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhao, Y.; Xu, S.; Zhang, Z.; Xu, Y.; Zhang, J.; Chong, K. OsMADS57 together with OsTB1 coordinates transcription of its target OsWRKY94 and D14 to switch its organogenesis to defense for cold adaptation in rice. New Phytol. 2018, 218, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, R.; Zheng, M.; Liu, X.; Meng, F.; Wu, H.; Yao, Y.; Xin, M.; Peng, H.; Ni, Z.; et al. TaWRKY51 promotes lateral root formation through negative regulation of ethylene biosynthesis in wheat (Triticum aestivum L.). Plant J. 2018, 96, 372–388. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Shim, Y.; Gi, E.; Lee, B.; An, G.; Kang, K.; Paek, N. The MYB-related transcription factor RADIALIS-LIKE3 (OsRL3) functions in ABA-induced leaf senescence and salt sensitivity in rice. Environ. Exp. Bot. 2018, 156, 86–95. [Google Scholar] [CrossRef]
- Nan, H.; Gao, L. Genome-Wide Analysis of WRKY Genes and Their Response to Hormone and Mechanic Stresses in Carrot. Front. Genet. 2019, 10, 363. [Google Scholar] [CrossRef]
- Zhu, D.; Hou, L.; Xiao, P.; Guo, Y.; Deyholos, M.K.; Liu, X. VvWRKY30, a grape WRKY transcription factor, plays a positive regulatory role under salinity stress. Plant Sci. 2019, 280, 132–142. [Google Scholar] [CrossRef]
- Takahashi, N.; Ogita, N.; Takahashi, T.; Taniguchi, S.; Tanaka, M.; Seki, M.; Umeda, M. A regulatory module controlling stress-induced cell cycle arrest in Arabidopsis. eLife 2019, 8, e43944. [Google Scholar] [CrossRef]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Shao, C.; Bao, Z.; Liu, G.; Bao, M. Single-repeat R3 MYB transcription factors from Platanus acerifolia negatively regulate trichome formation in Arabidopsis. Planta 2019, 249, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.; Song, S.; Ren, Q.; Wu, D.; Huang, H.; Chen, Y.; Fan, M.; Peng, W.; Ren, C.; Xie, D. The Jasmonate-ZIM-domain proteins interact with the WD-Repeat/bHLH/MYB complexes to regulate Jasmonate-mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana. Plant Cell 2011, 23, 1795–1814. [Google Scholar] [CrossRef] [PubMed]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Chao, Y.C.; Tseng, T.W.; Huang, C.K.; Lo, P.C.; Lu, C.A. Two MYB-related transcription factors play opposite roles in sugar signaling in Arabidopsis. Plant Mol. Biol. 2017, 93, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.; Guangyuan, H.; Guangxiao, Y.; Hussain, J.; Xu, Y. AP2/ERF Transcription Factor in Rice: Genome-Wide Canvas and Syntenic Relationships between Monocots and Eudicots. Evol. Bioinform. Online 2012, 8, 321–355. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Suzuki, K.; Fujimura, T.; Shinshi, H. Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol. 2006, 140, 411–432. [Google Scholar] [CrossRef] [PubMed]
- Shimono, M.; Sugano, S.; Nakayama, A.; Jiang, C.; Ono, K.; Toki, S.; Takatsuji, H. Rice WRKY45 Plays a Crucial Role in Benzothiadiazole-Inducible Blast Resistance. Plant Cell 2007, 19, 2064–2076. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Seemann, J.R.; Neuman, D.; Shen, Q.J. A WRKY Gene from Creosote Bush Encodes an Activator of the Abscisic Acid Signaling Pathway. J. Biol. Chem. 2004, 279, 55770–55779. [Google Scholar] [CrossRef]
- Li, W.; Pang, S.; Lu, Z.; Jin, B. Function and Mechanism of WRKY Transcription Factors in Abiotic Stress Responses of Plants. Plants 2020, 9, 1515. [Google Scholar] [CrossRef]
- Sahebi, M.; Hanafi, M.M.; Rafii, M.Y.; Mahmud, T.; Azizi, P.; Osman, M.; Abiri, R.; Taheri, S.; Kalhori, N.; Shabanimofrad, M.; et al. Improvement of Drought Tolerance in Rice (Oryza sativa L.): Genetics, Genomic Tools, and the WRKY Gene Family. BioMed Res. Int. 2018, 2018, 3158474. [Google Scholar] [CrossRef]
- Wang, C.T.; Ru, J.N.; Liu, Y.W.; Li, M.; Zhao, D.; Yang, J.F.; Fu, J.D.; Xu, Z.S. Maize WRKY Transcription Factor ZmWRKY106 Confers Drought and Heat Tolerance in Transgenic Plants. Int. J. Mol. Sci. 2018, 19, 3046. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.Y.; Du, Y.T.; Ma, J.; Min, D.H.; Jin, L.G.; Chen, J.; Chen, M.; Zhou, Y.B.; Ma, Y.Z.; Xu, Z.S.; et al. The WRKY Transcription Factor GmWRKY12 Confers Drought and Salt Tolerance in Soybean. Int. J. Mol. Sci. 2018, 19, 4087. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.F.; Liu, J.K.; Yang, F.M.; Zhang, G.Y.; Wang, D.; Zhang, L.; Ou, Y.B.; Yao, Y.A. The WRKY transcription factor WRKY8 promotes resistance to pathogen infection and mediates drought and salt stress tolerance in Solanum lycopersicum. Physiol. Plant 2020, 168, 98–117. [Google Scholar] [CrossRef] [PubMed]
- Villano, C.; Esposito, S.; D’Amelia, V.; Garramone, R.; Alioto, D.; Zoina, A.; Aversano, R.; Carputo, D. WRKY genes family study reveals tissue-specific and stress-responsive TFs in wild potato species. Sci. Rep. 2020, 10, 7196. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Roychoudhury, A. WRKY proteins: Signaling and regulation of expression during abiotic stress responses. Sci. World J. 2015, 2015, 807560. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Ma, S.; Ye, N.; Jiang, M.; Cao, J.; Zhang, J. WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 2017, 59, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kong, X.; Yang, L.; Fu, M.; Zhang, S. Genome-Wide Identification of WRKY Family Genes and the Expression Profiles in Response to Nitrogen Deficiency in Poplar. Genes 2022, 13, 2324. [Google Scholar] [CrossRef]
- Martín-Trillo, M.; Cubas, P. TCP genes: A family snapshot ten years later. Trends Plant Sci. 2010, 15, 31–39. [Google Scholar] [CrossRef]
- Craig, K.L.; Tyers, M. The F-box: A new motif for ubiquitin dependent proteolysis in cell cycle regulation and signal transduction. Prog. Biophys. Mol. Biol. 1999, 72, 299–328. [Google Scholar] [CrossRef]
- Singh, S.; Cornilescu, C.C.; Tyler, R.C.; Cornilescu, G.; Tonelli, M.; Lee, M.S.; Markley, J.L. Solution structure of a late embryogenesis abundant protein (LEA14) from Arabidopsis thaliana, a cellular stress-related protein. Protein Sci. 2005, 14, 2601–2609. [Google Scholar] [CrossRef]
- Wu, X.; Liu, T.; Zhang, Y.; Duan, F.; Neuhauser, B.; Ludewig, U.; Schulze, W.X.; Yuan, L. Ammonium and nitrate regulate NH4+ uptake activity of Arabidopsis ammonium transporter AtAMT1;3 via phosphorylation at multiple C-terminal sites. J. Exp. Bot. 2019, 70, 4919–4930. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Huang, X.; Hao, Y.; Su, W.; Liu, H.; Sun, G.; Chen, R.; Song, S. Ammonium Transporter (BcAMT1.2) Mediates the Interaction of Ammonium and Nitrate in Brassica campestris. Front. Plant Sci. 2019, 10, 1776. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Wang, Y.; Li, Y.; Li, Y.; Tan, P.; Wu, Z.; Shi, W.; Song, Z. Cloning and Functional Determination of Ammonium Transporter PpeAMT3;4 in Peach. BioMed Res. Int. 2020, 2020, 2147367. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Li, Y.; Lim, E.; Bowles, D.J. Higher plant glycosyltransferases. Genome Biol. 2001, 2, s3001–s3004. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.; Zhang, M.; Jia, B.; Heng, W.; Ye, Z.; Zhu, L.; Xu, X. Transcriptome sequencing analysis of two different genotypes of Asian pear reveals potential drought stress genes. Tree Genet. Genomes 2018, 14, 40. [Google Scholar] [CrossRef]
- Li, Y.; Xu, B.; Du, Q.; Zhang, D. Association genetics and expression patterns of a CBF4 homolog in Populus under abiotic stress. Mol. Genet. Genom. 2015, 290, 913–928. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ID | No. of Raw Reads (107) | No. of Clean Reads (107) | No. of Clean Basepairs (106) | No. of Mapped Reads (107) | Uniquely Mapped (107) | Mapped Percentage (%) |
|---|---|---|---|---|---|---|---|
| ZQUA44 | T1_1 | 9.49 | 9.18 | 13.55 | 5.13 | 5.00 | 54.48 |
| ZQUA44 | T1_2 | 9.06 | 8.78 | 13.11 | 4.77 | 4.63 | 52.76 |
| ZQUA44 | T1_3 | 9.73 | 9.49 | 14.24 | 4.09 | 3.94 | 41.53 |
| ZQUA44 | T2_1 | 9.60 | 9.28 | 13.67 | 4.91 | 4.79 | 51.58 |
| ZQUA44 | T2_2 | 9.35 | 9.08 | 13.57 | 3.79 | 3.62 | 39.82 |
| ZQUA44 | T2_3 | 10.47 | 10.11 | 15.17 | 5.46 | 5.32 | 52.66 |
| ZQUA44 | CK_1 | 10.28 | 9.89 | 14.85 | 5.72 | 5.57 | 56.27 |
| ZQUA44 | CK_2 | 9.18 | 8.84 | 13.07 | 4.51 | 4.33 | 48.91 |
| ZQUA44 | CK_3 | 8.81 | 8.58 | 12.82 | 3.63 | 3.50 | 40.75 |
| ZQUB15 | T1_1 | 9.83 | 9.51 | 14.05 | 4.09 | 3.93 | 41.35 |
| ZQUB15 | T1_2 | 9.70 | 9.34 | 13.95 | 4.70 | 4.56 | 48.81 |
| ZQUB15 | T1_3 | 8.42 | 8.15 | 12.21 | 3.37 | 3.26 | 40.01 |
| ZQUB15 | T2_1 | 9.58 | 9.22 | 13.59 | 4.99 | 4.87 | 52.75 |
| ZQUB15 | T2_2 | 8.76 | 8.52 | 12.73 | 3.36 | 3.24 | 37.98 |
| ZQUB15 | T2_3 | 10.58 | 10.30 | 15.37 | 4.30 | 4.15 | 40.34 |
| ZQUB15 | CK_1 | 8.47 | 8.22 | 12.27 | 3.21 | 3.07 | 37.37 |
| ZQUB15 | CK_2 | 8.97 | 8.68 | 12.96 | 4.29 | 4.19 | 48.24 |
| ZQUB15 | CK_3 | 11.11 | 10.72 | 16.04 | 5.11 | 4.99 | 46.64 |
| Groups of DEGs | GO Term | Description | Number of Enriched DEGs | p-Value | FDR |
|---|---|---|---|---|---|
| G1 | GO:0016701 | oxidoreductase activity, acting on single donors with incorporation of molecular oxygen | 5 | 4.00 × 10−6 | 0.001 |
| G1 | GO:0003824 | catalytic activity | 80 | 0.0003 | 0.039 |
| G1 | GO:0016757 | transferase activity, transferring glycosyl groups | 11 | 0.00045 | 0.039 |
| G1 | GO:0016563 | transcription activator activity | 6 | 0.00074 | 0.048 |
| G2 | GO:0016491 | oxidoreductase activity | 7 | 1.90 × 10−5 | 0.0001 |
| G2 | GO:0003824 | catalytic activity | 13 | 0.0015 | 0.0042 |
| G3 | GO:0003824 | catalytic activity | 66 | 7.00 × 10−12 | 1.20 × 10−9 |
| G3 | GO:0016787 | hydrolase activity | 27 | 1.10 × 10−5 | 0.00094 |
| G3 | GO:0016491 | oxidoreductase activity | 16 | 1.80 × 10−5 | 0.0011 |
| G3 | GO:0004091 | carboxylesterase activity | 8 | 3.40 × 10−5 | 0.0015 |
| AT | GO:0003824 | catalytic activity | 96 | 8.50 × 10−8 | 2.40 × 10−5 |
| AT | GO:0016491 | oxidoreductase activity | 25 | 4.90 × 10−6 | 0.00069 |
| AT | GO:0005215 | transporter activity | 22 | 0.00014 | 0.013 |
| AT | GO:0046527 | glucosyltransferase activity | 6 | 0.00024 | 0.017 |
| AT | GO:0022892 | substrate- transporter activity | 17 | 0.00035 | 0.018 |
| AT | GO:0016706 | oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, 2-oxoglutarate as one donor, and incorporation of one atom each of oxygen into both donors | 5 | 0.00037 | 0.018 |
| AT | GO:0022857 | transmembrane transporter activity | 17 | 0.0007 | 0.028 |
| AT | GO:0016705 | oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen | 6 | 0.0016 | 0.047 |
| AT | GO:0022891 | substrate- transmembrane transporter activity | 14 | 0.0017 | 0.047 |
| AT | GO:0016758 | transferase activity, transferring hexosyl groups | 8 | 0.0014 | 0.047 |
| BT | GO:0003824 | catalytic activity | 84 | 2.00 × 10−8 | 4.00 × 10−6 |
| BT | GO:0016491 | oxidoreductase activity | 22 | 5.90 × 10−6 | 0.0006 |
| BT | GO:0008194 | UDP-glycosyltransferase activity | 8 | 2.10 × 10−5 | 0.0014 |
| BT | GO:0016758 | transferase activity, transferring hexosyl groups | 9 | 7.20 × 10−5 | 0.0037 |
| BT | GO:0016757 | transferase activity, transferring glycosyl groups | 10 | 0.00052 | 0.021 |
| BT | GO:0046527 | glucosyltransferase activity | 5 | 0.00074 | 0.025 |
| BT | GO:0016209 | antioxidant activity | 5 | 0.0011 | 0.033 |
| BT | GO:0016765 | transferase activity, transferring alkyl or aryl (other than methyl) groups | 5 | 0.0014 | 0.036 |
| Groups | KEGG Pathway | ID | DEGs Number | p-Value | Q-Value |
|---|---|---|---|---|---|
| G1 | Ascorbate and aldarate metabolism | ath00053 | 4 | 0.0002 | 0.0085 |
| G1 | Arginine and proline metabolism | ath00330 | 3 | 0.0024 | 0.0547 |
| G1 | Tyrosine metabolism | ath00350 | 2 | 0.0164 | 0.2424 |
| G1 | Galactose metabolism | ath00052 | 2 | 0.0260 | 0.2424 |
| G1 | Alanine, aspartate and glutamate metabolism | ath00250 | 2 | 0.0298 | 0.2424 |
| G1 | Limonene and pinene degradation | ath00903 | 1 | 0.0346 | 0.2424 |
| G1 | Monoterpenoid biosynthesis | ath00902 | 1 | 0.0377 | 0.2424 |
| G1 | Metabolic pathways | ath01100 | 17 | 0.0439 | 0.2471 |
| G2 | Phenylpropanoid biosynthesis | ath00940 | 2 | 0.0040 | 0.0202 |
| G2 | Sulfur metabolism | ath00920 | 1 | 0.0249 | 0.0623 |
| G3 | Phenylalanine, tyrosine and tryptophan biosynthesis | ath00400 | 2 | 0.0111 | 0.1867 |
| G3 | Stilbenoid, diarylheptanoid and gingerol biosynthesis | ath00945 | 1 | 0.0243 | 0.1867 |
| G3 | Metabolic pathways | ath01100 | 12 | 0.02515 | 0.1867 |
| G3 | Glutathione metabolism | ath00480 | 2 | 0.0325 | 0.1867 |
| AS | Galactose metabolism | ath00052 | 3 | 0.0030 | 0.0938 |
| AS | Plant hormone signal transduction | ath04075 | 6 | 0.0063 | 0.0938 |
| AS | Protein processing in endoplasmic reticulum | ath04141 | 5 | 0.0072 | 0.0938 |
| AS | Metabolic pathways | ath01100 | 21 | 0.0112 | 0.1088 |
| AS | Nitrogen metabolism | ath00910 | 2 | 0.0288 | 0.1991 |
| AS | Cysteine and methionine metabolism | ath00270 | 3 | 0.0306 | 0.1991 |
| AS | Glycerolipid metabolism | ath00561 | 2 | 0.0414 | 0.2308 |
| BS | Cutin, suberine and wax biosynthesis | ath00073 | 2 | 0.0076 | 0.1249 |
| BS | Zeatin biosynthesis | ath00908 | 2 | 0.0109 | 0.1249 |
| Gene ID | AT1 (FPKM) | AT2 (FPKM) | ACK (FPKM) | BT1 (FPKM) | BT2 (FPKM) | BCK (FPKM) | Uniprot | Symbol | Subcluster |
|---|---|---|---|---|---|---|---|---|---|
| Eucgr.A01486 | 0.64 | 0.87 | 6.35 | 4.08 | 2.92 | 4.94 | Q5SN75 | P2C08 | sub4 |
| Eucgr.A02858 | 0.24 | 0.30 | 9.65 | 10.76 | 1.75 | 3.41 | Q9FLI3 | P2C75 | sub4 |
| Eucgr.C03732 | 21.00 | 24.44 | 76.28 | 46.38 | 23.80 | 40.61 | P49597 | P2C56 | sub1 |
| Eucgr.F00253 | 0.55 | 0.51 | 0.00 | 0.70 | 0.00 | 0.00 | Q9FX08 | P2C12 | sub6 |
| Eucgr.H04087 | 16.26 | 226.00 | 35.9 | 19.74 | 15.81 | 14.61 | Q3EAF9 | P2C49 | sub1 |
| Eucgr.J02003 | 8.73 | 181.00 | 83.02 | 56.76 | 26.57 | 60.24 | Q9ZW21 | P2C24 | sub4 |
| Eucgr.C03337 | 1222.61 | 1033.70 | 638.04 | 37.29 | 57.41 | 105.58 | Q9ZRA4 | AB19A | sub3 |
| Eucgr.C03536 | 1079.85 | 930.58 | 667.89 | 227.95 | 439.95 | 6177.00 | Q9ZRA4 | AB19A | sub3 |
| Eucgr.D00606 | 58.97 | 124.94 | 334.92 | 267.52 | 238.79 | 182.00 | Q05349 | 12KD | sub4 |
| Eucgr.I01276 | 3.24 | 3.83 | 1.87 | 7.70 | 10.80 | 26.74 | Q6NMM0 | SAU61 | sub4 |
| Eucgr.F03208 | 38.00 | 1.29 | 0.67 | 2.78 | 0.74 | 0.95 | Q9SQ80 | G2OX1 | sub6 |
| Eucgr.F04125 | 22.90 | 32.45 | 8.65 | 10.03 | 4.30 | 3.94 | P46687 | GASA3 | sub3 |
| Eucgr.K02472 | 20.46 | 20.90 | 18.74 | 117.00 | 41.92 | 58.02 | Q6NMQ7 | GASA6 | sub5 |
| Eucgr.F00192 | 13.57 | 17.63 | 27.34 | 38.65 | 23.74 | 14.97 | Q8LC30 | RAP21 | sub1 |
| Eucgr.F02317 | 1.08 | 0.47 | 1.55 | 58.00 | 1.66 | 0.87 | O22174 | ERF08 | sub1 |
| Eucgr.F02691 | 0.04 | 0.36 | 23.00 | 34.00 | 0.00 | 0.19 | Q70II3 | EF110 | sub1 |
| Eucgr.H01659 | 0.02 | 0.05 | 0.53 | 1.38 | 0.00 | 0.00 | Q9SZ06 | EF109 | sub1 |
| Eucgr.K00128 | 19.27 | 9.67 | 16.3 | 75.95 | 18.5 | 13.04 | Q9LY05 | EF106 | sub1 |
| Eucgr.F04203 | 0.13 | 0.20 | 8.69 | 4.30 | 0.27 | 0.15 | Q9FGF8 | ABR1 | sub4 |
| Eucgr.G01970 | 0.15 | 0.37 | 3.51 | 6.09 | 1.74 | 0.32 | Q9LYU3 | EF113 | sub4 |
| Eucgr.H03965 | 5.20 | 12.24 | 20.35 | 18.47 | 21.78 | 6.23 | P42736 | RAP23 | sub4 |
| Eucgr.C04221 | 30.5 | 24.82 | 14.00 | 22.97 | 11.44 | 29.80 | Q9XI33 | WIN1 | sub6 |
| Eucgr.F02319 | 7.73 | 5.56 | 7.53 | 12.26 | 6.01 | 2.54 | Q8LC30 | RAP21 | sub6 |
| Eucgr.I00422 | 12.92 | 11.51 | 3.99 | 14.41 | 20.31 | 13.61 | O65665 | ERF60 | sub6 |
| Eucgr.K00126 | 69.17 | 62.26 | 47.43 | 89.37 | 59.17 | 42.78 | Q8VY90 | EF105 | sub6 |
| Eucgr.A01146 | 18.41 | 11.39 | 6.04 | 13.68 | 12.62 | 5.29 | Q8L8B8 | LOG3 | sub6 |
| Eucgr.B02321 | 5.67 | 7.47 | 35.5 | 11.99 | 13.25 | 6.79 | O81077 | ABAH2 | sub1 |
| Eucgr.C01524 | 7.03 | 4.49 | 2.78 | 23.39 | 14.27 | 12.84 | Q9SKK0 | EBF1 | sub1 |
| Eucgr.C03157 | 33.47 | 38.36 | 13.15 | 25.79 | 30.28 | 12.39 | Q9FUJ1 | CKX7 | sub6 |
| Eucgr.E01149 | 13.82 | 13.43 | 23.66 | 19.39 | 9.18 | 6.47 | Q949P1 | ABAH1 | sub1 |
| Eucgr.G01437 | 0.04 | 0.29 | 1.74 | 6.04 | 1.81 | 1.43 | Q9LJK2 | ABAH4 | sub1 |
| Eucgr.G03093 | 3.61 | 1.72 | 1.54 | 3.81 | 0.47 | 0.53 | Q8S8E3 | PYL6 | sub6 |
| Eucgr.I01127 | 15.24 | 21.06 | 9.75 | 27.58 | 26.97 | 8.78 | Q8W3P8 | AOG | sub1 |
| Eucgr.I01201 | 0.14 | 0.61 | 0.11 | 0.42 | 3.41 | 3.39 | Q6RYA0 | SABP2 | sub4 |
| Eucgr.J00169 | 6.76 | 4.55 | 2.08 | 5.09 | 1.23 | 0.54 | O80920 | PYL4 | sub6 |
| Eucgr.K02472 | 20.46 | 20.9 | 18.74 | 11.17 | 41.92 | 58.02 | Q6NMQ7 | GASA6 | sub5 |
| Eucgr.B03374 | 1.38 | 2.09 | 4.49 | 1.70 | 1.41 | 0.96 | Q9ZWS9 | ARR3 | sub1 |
| Eucgr.B02620 | 11.64 | 26.09 | 57.13 | 113.34 | 90.20 | 95.91 | Q39182 | DEF02 | sub2 |
| Eucgr.H05052 | 223.79 | 163.36 | 302.39 | 893.65 | 3003.64 | 3268.99 | Q07502 | DEF | sub4 |
| Transcription Factors | G1 | G2 | G3 | AS | BS |
|---|---|---|---|---|---|
| C2H2 | 1 | 1 | |||
| CPP | 1 | ||||
| ERF | 7 | 1 | 2 | ||
| MYB | 4 | 3 | 1 | 2 | |
| NAC | 6 | 1 | 1 | ||
| RAV | 1 | ||||
| WRKY | 2 | 5 | |||
| B3 | 3 | ||||
| bHLH | 3 | 1 | 1 | ||
| bZIP | 2 | ||||
| C3H | 3 | 2 | |||
| Dof | 2 | ||||
| G2-like | 3 | ||||
| HD-ZIP | 1 | 1 | |||
| HSF | 1 | 1 | 2 | ||
| MYB_related | 1 | 3 | 2 | ||
| NF-YA | 1 | ||||
| TCP | 1 | ||||
| MIKC_MADS | 1 | ||||
| LBD | 1 | ||||
| SBP | 1 |
| Groups of DEGs | Gene ID | Gene Names | T1 | T2 | CK | log2FPKM (T1/CK) | p Value | log2FPKM (T2/CK) | p Value |
|---|---|---|---|---|---|---|---|---|---|
| AT | Eucgr.H00996 | WRKY23 | 1.57 | 4.82 | 8.19 | −2.38 | 0.00 | −0.76 | 0.18 |
| AT | Eucgr.B03520 | WRKY75 | 6.93 | 9.67 | 24.10 | −1.80 | 0.00 | −1.32 | 0.03 |
| BT | Eucgr.B04010 | WRKY26 | 15.39 | 8.77 | 4.79 | 1.68 | 0.00 | 0.87 | 0.07 |
| BT | Eucgr.K02940 | WRKY33 | 8.01 | 5.62 | 2.84 | 1.50 | 0.00 | 0.99 | 0.06 |
| BT | Eucgr.C00675 | WRKY50 | 26.21 | 17.53 | 7.47 | 1.81 | 0.00 | 1.23 | 0.02 |
| BT | Eucgr.E04011 | WRKY6 | 4.21 | 2.48 | 1.07 | 1.98 | 0.00 | 1.22 | 0.08 |
| BT | Eucgr.I01633 | WRKY75 | 1.72 | 4.70 | 0.54 | 1.66 | 0.16 | 3.11 | 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Xu, F.; Pan, W.; Zhang, W.; Liao, H.; Zhu, B.; Xu, B.; Chen, X.; Yang, H. Comparative Transcriptome Analysis of High- and Low-Growth Genotypes of Eucalyptus urophylla in Response to Long-Term Nitrogen Deficiency. Genes 2024, 15, 60. https://doi.org/10.3390/genes15010060
Yang X, Xu F, Pan W, Zhang W, Liao H, Zhu B, Xu B, Chen X, Yang H. Comparative Transcriptome Analysis of High- and Low-Growth Genotypes of Eucalyptus urophylla in Response to Long-Term Nitrogen Deficiency. Genes. 2024; 15(1):60. https://doi.org/10.3390/genes15010060
Chicago/Turabian StyleYang, Xiaohui, Fang Xu, Wen Pan, Weihua Zhang, Huanqin Liao, Baozhu Zhu, Bin Xu, Xinyu Chen, and Huixiao Yang. 2024. "Comparative Transcriptome Analysis of High- and Low-Growth Genotypes of Eucalyptus urophylla in Response to Long-Term Nitrogen Deficiency" Genes 15, no. 1: 60. https://doi.org/10.3390/genes15010060
APA StyleYang, X., Xu, F., Pan, W., Zhang, W., Liao, H., Zhu, B., Xu, B., Chen, X., & Yang, H. (2024). Comparative Transcriptome Analysis of High- and Low-Growth Genotypes of Eucalyptus urophylla in Response to Long-Term Nitrogen Deficiency. Genes, 15(1), 60. https://doi.org/10.3390/genes15010060