


KMT2D Deficiency Causes Sensorineural Hearing Loss in Mice and Humans

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Auditory Brainstem Response and Distortion Product Otoacoustic Emission Procedure
2.3. Statistical Analysis
2.4. Immunofluorescence
2.5. Volumetric Measurements
2.6. Participants
3. Results
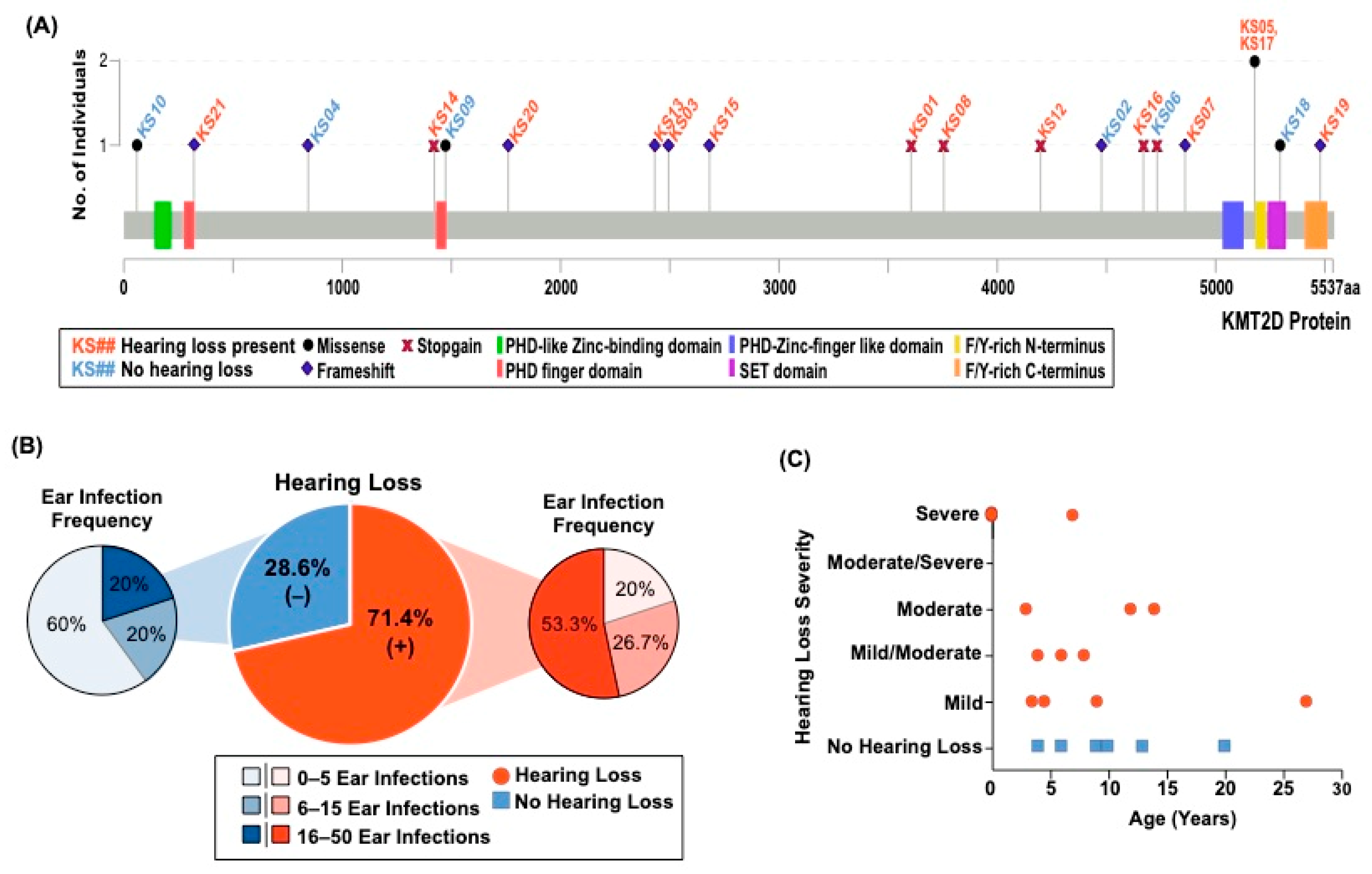
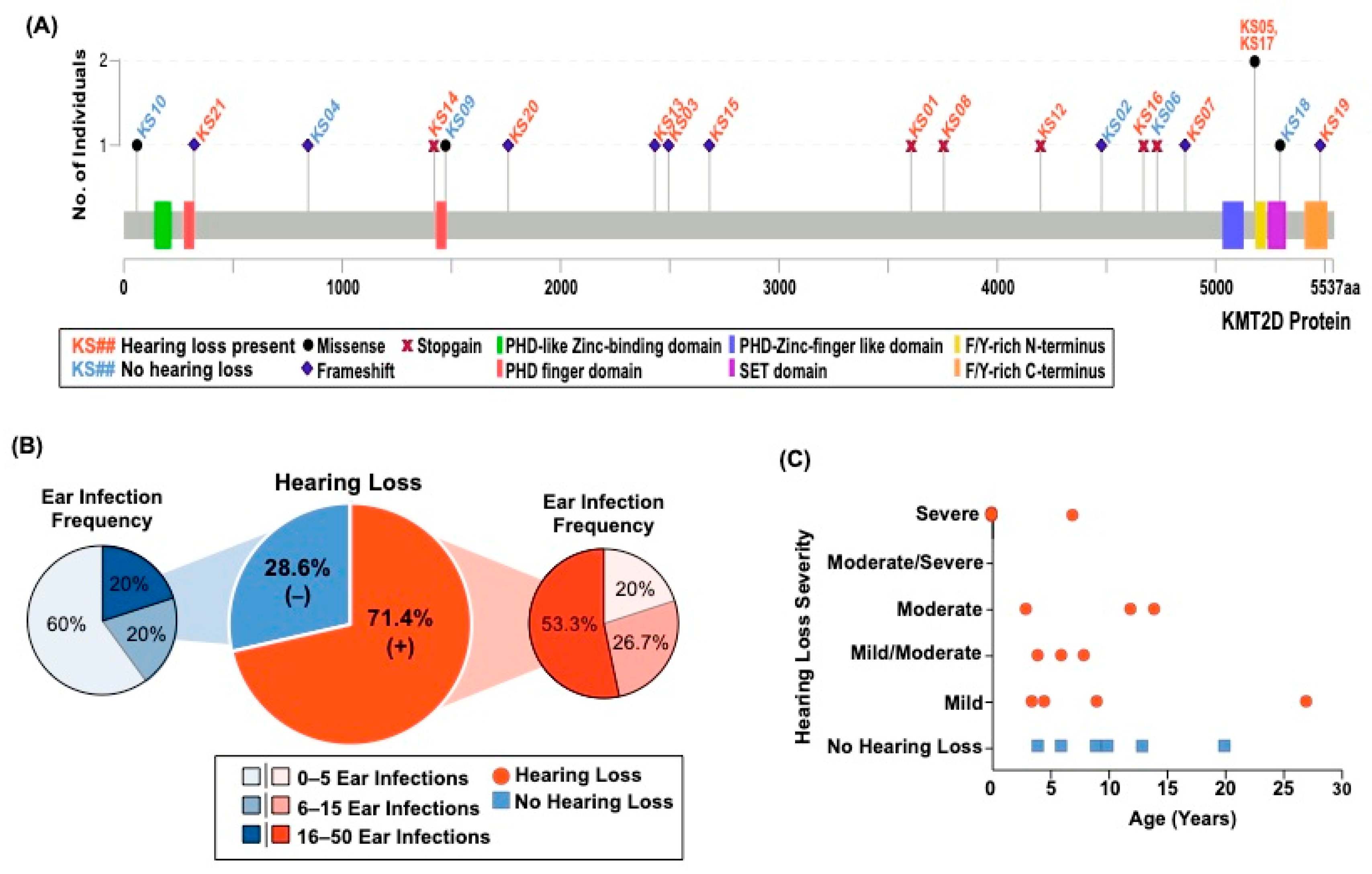
3.1. Individuals with KS1 Have Multiple Factors Contributing to Hearing Loss
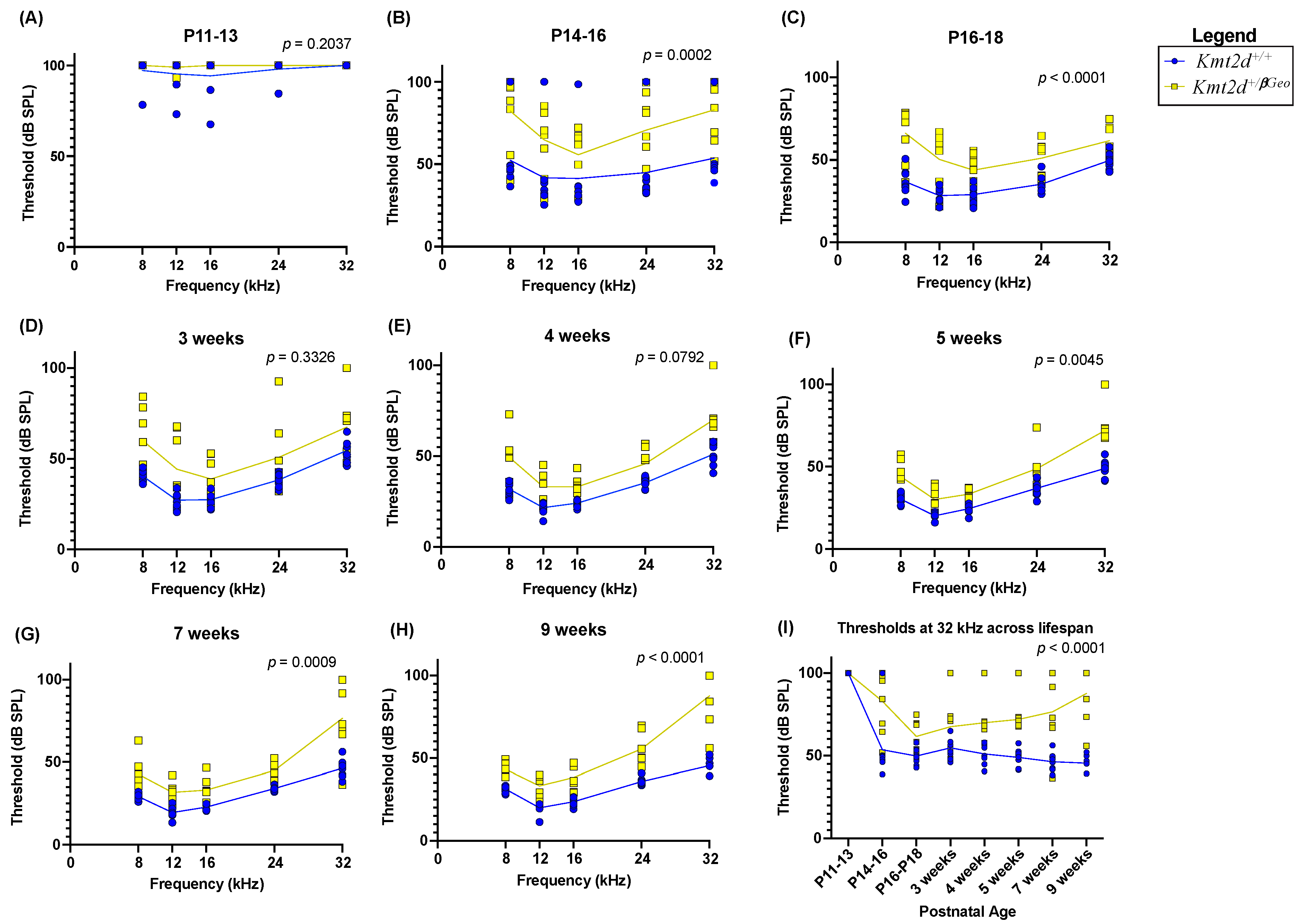
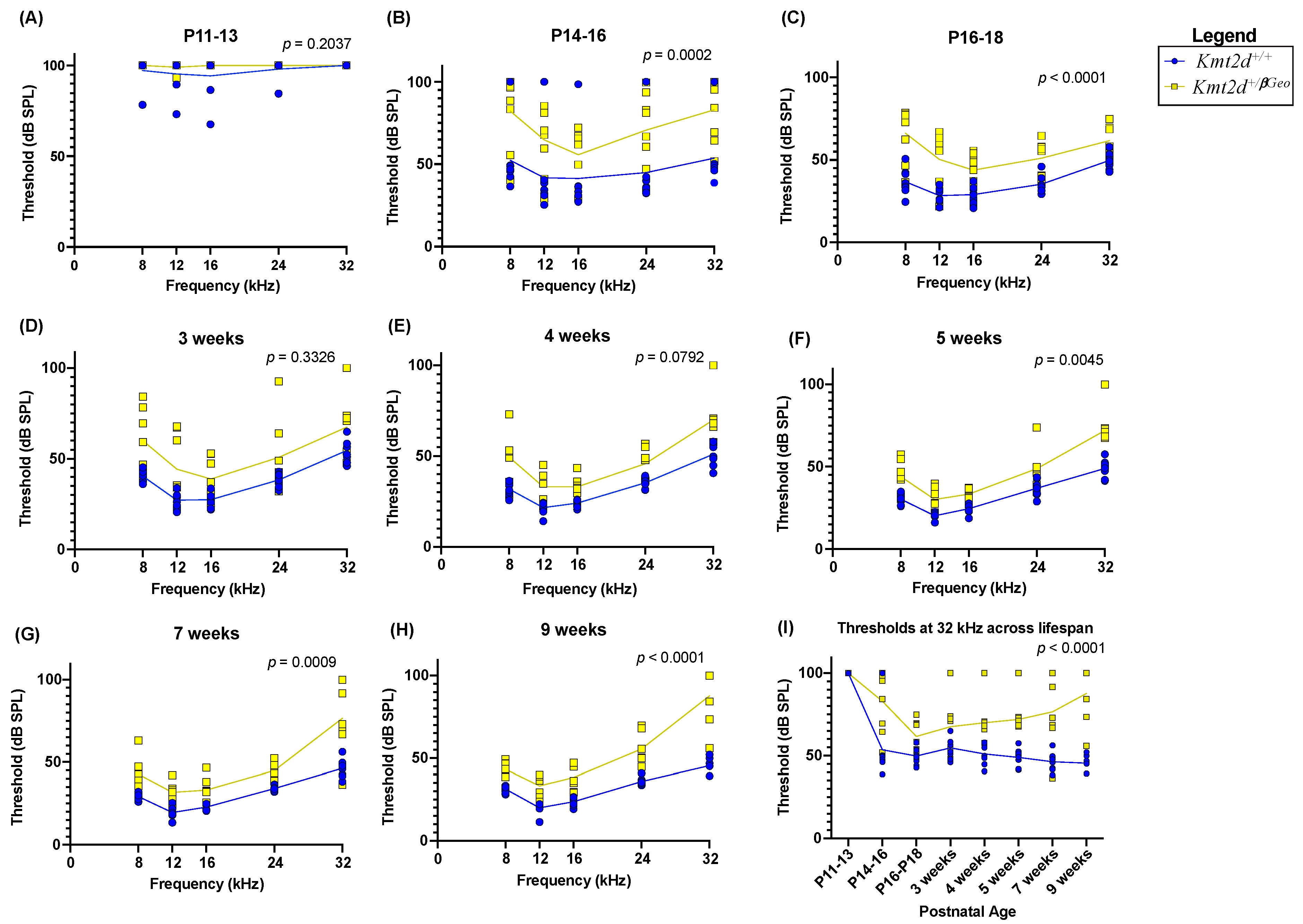
3.2. Kmt2d+/βGeo Mice Have Early Hearing Impairment
3.3. Hearing Loss in Kmt2d+/βGeo Mice Is Exacerbated with Age
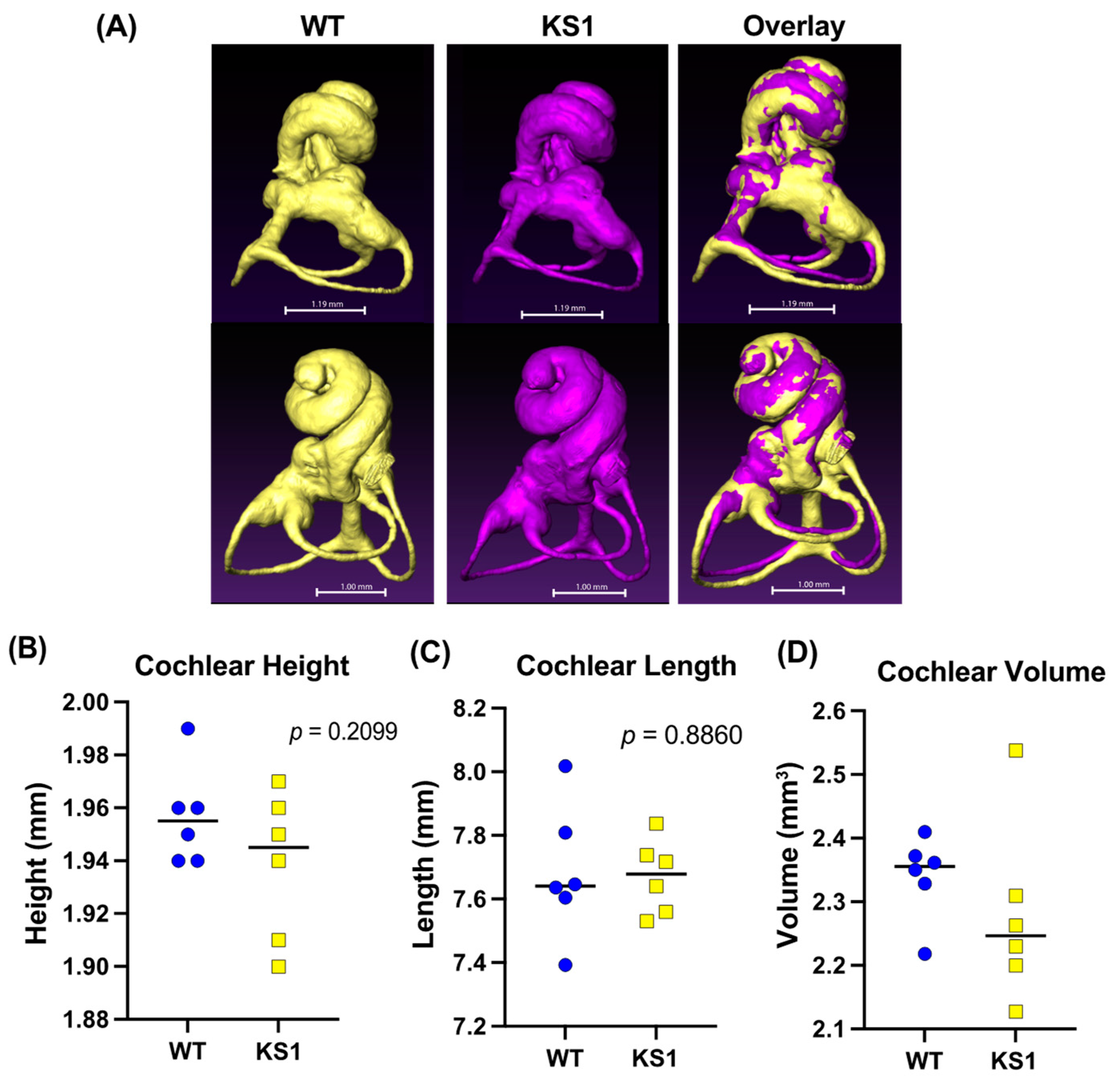
3.4. Kmt2d+/βGeo Mice Have No Obvious Structural Abnormalities in the Inner Ear
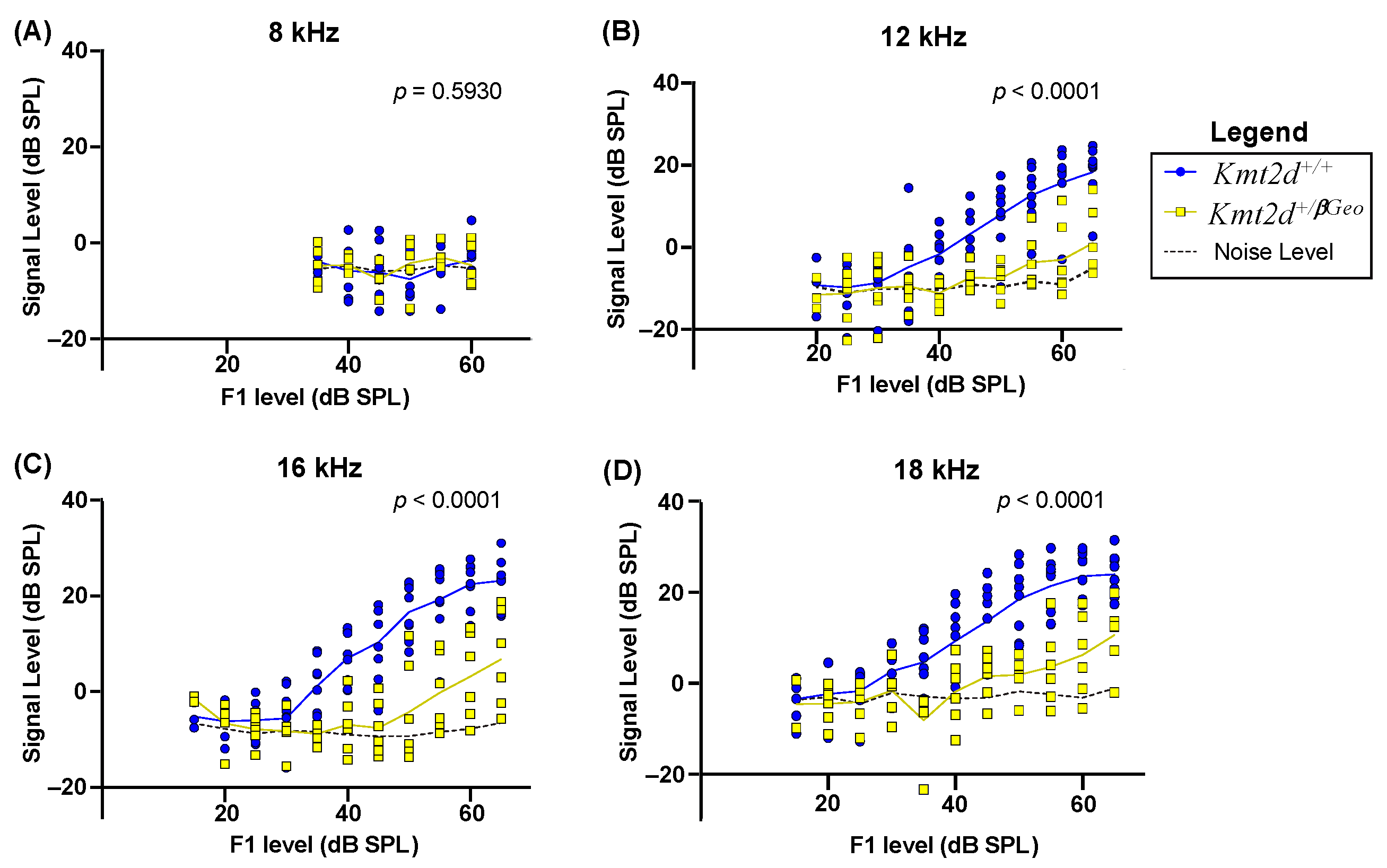
3.5. Kmt2d+/βGeo Mice Display Diminished DPOAE Levels
3.6. Outer Hair Cells Have Subtle Damage in Adult Kmt2d+/βGeo Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fahrner, J.A.; Bjornsson, H.T. Mendelian disorders of the epigenetic machinery: Postnatal malleability and therapeutic prospects. Hum. Mol. Genet. 2019, 28, R254–R264. [Google Scholar] [CrossRef] [PubMed]
- Gopinathan, G.; Diekwisch, T.G.H. Epigenetics and Early Development. J. Dev. Biol. 2022, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Sartorelli, V.; Puri, P.L. Shaping Gene Expression by Landscaping Chromatin Architecture: Lessons from a Master. Mol. Cell 2018, 71, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Chen, L.H.; Huang, Y.; Chang, C.; Wang, P.; Pirozzi, C.J.; Qin, X.; Bao, X.; Greer, P.K.; McLendon, R.E.; et al. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget 2013, 4, 2144–2153. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sharma, A.; Dhar, S.S.; Lee, S.; Gu, B.; Chan, C.; Lin, H.; Lee, M.G. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014, 74, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Marwaha, A.; Costain, G.; Cytrynbaum, C.; Mendoza-Londono, R.; Chad, L.; Awamleh, Z.; Chater-Diehl, E.; Choufani, S.; Weksberg, R. The utility of DNA methylation signatures in directing genome sequencing workflow: Kabuki syndrome and CDK13-related disorder. Am. J. Med. Genet. Part A 2022, 188, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Butcher, D.T.; Cytrynbaum, C.; Turinsky, A.L.; Siu, M.T.; Inbar-Feigenberg, M.; Mendoza-Londono, R.; Chitayat, D.; Walker, S.; Machado, J.; Caluseriu, O.; et al. CHARGE and Kabuki Syndromes: Gene-Specific DNA Methylation Signatures Identify Epigenetic Mechanisms Linking These Clinically Overlapping Conditions. Am. J. Hum. Genet. 2017, 100, 773–788. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Rodenhiser, D.; Schwartz, C.; Sadikovic, B. The defining DNA methylation signature of Kabuki syndrome enables functional assessment of genetic variants of unknown clinical significance. Epigenetics 2017, 12, 923–933. [Google Scholar] [CrossRef]
- Froimchuk, E.; Jang, Y.; Ge, K. Histone H3 lysine 4 methyltransferase KMT2D. Gene 2017, 627, 337–342. [Google Scholar] [CrossRef]
- Niikawa, N.; Kuroki, Y.; Kajii, T.; Matsuura, N.; Ishikiriyama, S.; Tonoki, H.; Ishikawa, N.; Yamada, Y.; Fujita, M.; Umemoto, H.; et al. Kabuki make-up (Niikawa-Kuroki) syndrome: A study of 62 patients. Am. J. Med. Genet. 1988, 31, 565–589. [Google Scholar] [CrossRef] [PubMed]
- Barry, K.K.; Tsaparlis, M.; Hoffman, D.; Hartman, D.; Adam, M.P.; Hung, C.; Bodamer, O.A. From Genotype to Phenotype-A Review of Kabuki Syndrome. Genes 2022, 13, 1761. [Google Scholar] [CrossRef] [PubMed]
- Barozzi, S.; Di Berardino, F.; Atzeri, F.; Filipponi, E.; Cerutti, M.; Selicorni, A.; Cesarani, A. Audiological and vestibular findings in the Kabuki syndrome. Am. J. Med. Genet. A 2009, 149A, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Boniel, S.; Szymańska, K.; Śmigiel, R.; Szczałuba, K. Kabuki Syndrome-Clinical Review with Molecular Aspects. Genes 2021, 12, 468. [Google Scholar] [CrossRef] [PubMed]
- Urbančič, N.B.; Battelino, S.; Tesovnik, T.; Podkrajšek, K.T. The Importance of Early Genetic Diagnostics of Hearing Loss in Children. Medicina 2020, 56, 471. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.R.; Niparko, J.K.; Ferrucci, L. Hearing loss prevalence in the United States. Arch. Intern. Med. 2011, 171, 1851–1852. [Google Scholar] [CrossRef]
- Pilarowski, G.O.; Cazares, T.; Zhang, L.; Benjamin, J.S.; Liu, K.; Jagannathan, S.; Mousa, N.; Kasten, J.; Barski, A.; Lindsley, A.W.; et al. Abnormal Peyer patch development and B-cell gut homing drive IgA deficiency in Kabuki syndrome. J. Allergy Clin. Immunol. 2020, 145, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, J.A.; Lin, W.; Riddle, R.C.; Boukas, L.; DeLeon, V.B.; Chopra, S.; Lad, S.E.; Luperchio, T.R.; Hansen, K.D.; Bjornsson, H.T. Precocious chondrocyte differentiation disrupts skeletal growth in Kabuki syndrome mice. JCI Insight 2019, 4, e129380. [Google Scholar] [CrossRef]
- Hannibal, M.C.; Buckingham, K.J.; Ng, S.B.; Ming, J.E.; Beck, A.E.; McMillin, M.J.; Gildersleeve, H.I.; Bigham, A.W.; Tabor, H.K.; Mefford, H.C.; et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am. J. Med. Genet. A 2011, 155A, 1511–1516. [Google Scholar] [CrossRef]
- van Ravenswaaij-Arts, C.; Martin, D.M. New insights and advances in CHARGE syndrome: Diagnosis, etiologies, treatments, and research discoveries. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 397–406. [Google Scholar] [CrossRef]
- Patel, N.; Alkuraya, F.S. Overlap between CHARGE and Kabuki syndromes: More than an interesting clinical observation? Am. J. Med. Genet. A 2015, 167A, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Schulz, Y.; Freese, L.; Mänz, J.; Zoll, B.; Völter, C.; Brockmann, K.; Bögershausen, N.; Becker, J.; Wollnik, B.; Pauli, S. CHARGE and Kabuki syndromes: A phenotypic and molecular link. Hum. Mol. Genet. 2014, 23, 4396–4405. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.E.; Russell, K.L.; Bason, L.; McDonald-McGinn, D.M.; Zackai, E.H. Coloboma and other ophthalmologic anomalies in Kabuki syndrome: Distinction from charge association. Am. J. Med. Genet 2003, 123A, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Funamura, J.L. Evaluation and management of nonsyndromic congenital hearing loss. Curr. Opin. Otolaryngol. Head Neck Surg. 2017, 25, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, H.T.; Benjamin, J.S.; Zhang, L.; Weissman, J.; Gerber, E.E.; Chen, Y.; Vaurio, R.G.; Potter, M.C.; Hansen, K.D.; Dietz, H.C. Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci. Transl. Med. 2014, 6, 256ra135. [Google Scholar] [CrossRef] [PubMed]
- Early, S.; Du, E.; Boussaty, E.; Friedman, R. Genetics of noise-induced hearing loss in the mouse model. Hear. Res. 2022, 425. [Google Scholar] [CrossRef]
- Schrode, K.M.; Muniak, M.A.; Kim, Y.; Lauer, A.M. Central Compensation in Auditory Brainstem after Damaging Noise Exposure. eNeuro 2018, 5, ENEURO.0250-18.2018. [Google Scholar] [CrossRef]
- Schinedlin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji. an open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Bouxsein, M.L.; Boyd, S.K.; Christiansen, B.A.; Guldberg, R.E.; Jepsen, K.J.; Müller, R. Guidelines for assessment of bone microstructure in rodents using micro–computed tomography. J. Bone Miner. Res. 2010, 25, 1468–1486. [Google Scholar] [CrossRef]
- Song, L.; McGee, J.; Walsh, E.J. Frequency- and level-dependent changes in auditory brainstem responses (ABRS) in developing mice. J. Acoust. Soc. Am. 2006, 119, 2242–2257. [Google Scholar] [CrossRef]
- Mhatre, A.N.; Tajudeen, B.; Welt, E.M.; Wartmann, C.; Long, G.R.; Lalwani, A.K. Temporary reduction of distortion product otoacoustic emissions (DPOAEs) immediately following auditory brainstem response (ABR). Hear. Res. 2010, 269, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Aldè, M.; Cantarella, G.; Zanetti, D.; Pignataro, L.; La Mantia, I.; Maiolino, L.; Ferlito, S.; Di Mauro, P.; Cocuzza, S.; Lechien, J.R.; et al. Autosomal Dominant Non-Syndromic Hearing Loss (DFNA): A Comprehensive Narrative Review. Biomedicines 2023, 11, 1616. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.J.; Abrams, K.S.; Amburgey, K.N.; Wang, Y.; Henry, K.S. Effects of selective auditory-nerve damage on the behavioral audiogram and temporal integration in the budgerigar. Hear. Res. 2019, 374, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Lobarinas, E.; Salvi, R.; Ding, D. Insensitivity of the audiogram to carboplatin induced inner hair cell loss in chinchillas. Hear. Res. 2013, 302, 113–120. [Google Scholar] [CrossRef]
- Margot, H.; Boursier, G.; Duflos, C.; Sanchez, E.; Amiel, J.; Andrau, J.C.; Arpin, S.; Brischoux-Boucher, E.; Boute, O.; Burglen, L.; et al. Immunopathological manifestations in Kabuki syndrome: A registry study of 177 individuals. Genet. Med. 2020, 22, 181–188. [Google Scholar] [CrossRef]
- Faundes, V.; Goh, S.; Akilapa, R.; Bezuidenhout, H.; Bjornsson, H.T.; Bradley, L.; Brady, A.F.; Brischoux-Boucher, E.; Brunner, H.; Bulk, S.; et al. Clinical delineation, sex differences, and genotype-phenotype correlation in pathogenic KDM6A variants causing X-linked Kabuki syndrome type 2. Genet. Med. 2021, 23, 1202–1210. [Google Scholar] [CrossRef]
- Miyake, N.; Mizuno, S.; Okamoto, N.; Ohashi, H.; Shiina, M.; Ogata, K.; Tsurusaki, Y.; Nakashima, M.; Saitsu, H.; Niikawa, N.; et al. KDM6A Point Mutations Cause Kabuki Syndrome. Hum. Mutat. 2012, 34, 108–110. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, J.; Yin, L.; Pu, Y. TMT-Based Quantitative Proteomics Reveals Cochlear Protein Profile Alterations in Mice with Noise-Induced Hearing Loss. Int. J. Environ. Res. Public Health 2021, 19, 382. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Variant | Age | Hearing Loss/Type | Age of Hearing Loss | Severity | Total # of Ear Infections | Outer, Middle, and/or Inner Ear Abnormalities |
|---|---|---|---|---|---|---|---|
| KS01 | c.10813C>T | 22 | +/Bilateral | 14 | Moderate | 2 | - |
| KS02 | c.13432dupC | 20 | - | NA | NA | Many | - |
| KS03 | c.7481_7482insT | 20 | +/Bilateral | 4 | Mild/moderate | 20+ | + |
| KS04 | c.2533delC | 9 | - | NA | NA | 3 | - |
| KS05 | c.15536G>A | 30 | +/Unilateral | 27 | Mild | ~16 | - |
| KS06 | c.14194C>T | 4 | - | NA | NA | 1 | + |
| KS07 | c.14580dupT | 36 | +/Bilateral | 6 | Mild/moderate | ~40 | - |
| KS08 | c.11263C>T | 13 | +/Bilateral | 7 | Mild | 12 | - |
| KS09 | c.4421G>T | 10 | - | NA | NA | 9 | - |
| KS10 | c.175A>G | 6 | - | NA | NA | 1 | - |
| KS11 | IVS50+5G>A | 15 | +/Unilateral | 12 | Moderate | ~10 | NR |
| KS12 | c.12592C>T | 21 | +/Bilateral | 8 | Mild/moderate | 20 | + |
| KS13 | c.7291_7294delTCTG | 17 | +/Bilateral | At Birth | Profound | 1 | + |
| KS14 | c.4265G>A | 18 | +/Bilateral | 7 | Right-severe. Left consistent. | 30 | - |
| KS15 | c.8045_8046delAG | 3 | +/Bilateral | At Birth | Left- moderate/severe Right-severe | 10+ | - |
| KS16 | c.14006C>G | 29 | +/Bilateral | 3.5 | Mild | 25 | - |
| KS17 | c.15536G>A | 28 | +/Bilateral | 4.5 | 15–30 decibel hearing loss | ~50 | - |
| KS18 | c.15884G>C | 13 | -/Bilateral | 2 | Normal now but significant at 2 years of age | 8–9 | - |
| KS19 | c.16437delT | 15 | +/Bilateral | 3 | Right-moderate. Left-mild/moderate | 25+ | - |
| KS20 | c.5278_5279delAA | 12 | +/Unilateral | 9 | Mild | 8 | - |
| KS21 | c.970dupC | 1.7 | +/Bilateral | At birth | Profound | 0 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalinousky, A.J.; Luperchio, T.R.; Schrode, K.M.; Harris, J.R.; Zhang, L.; DeLeon, V.B.; Fahrner, J.A.; Lauer, A.M.; Bjornsson, H.T. KMT2D Deficiency Causes Sensorineural Hearing Loss in Mice and Humans. Genes 2024, 15, 48. https://doi.org/10.3390/genes15010048
Kalinousky AJ, Luperchio TR, Schrode KM, Harris JR, Zhang L, DeLeon VB, Fahrner JA, Lauer AM, Bjornsson HT. KMT2D Deficiency Causes Sensorineural Hearing Loss in Mice and Humans. Genes. 2024; 15(1):48. https://doi.org/10.3390/genes15010048
Chicago/Turabian StyleKalinousky, Allison J., Teresa R. Luperchio, Katrina M. Schrode, Jacqueline R. Harris, Li Zhang, Valerie B. DeLeon, Jill A. Fahrner, Amanda M. Lauer, and Hans T. Bjornsson. 2024. "KMT2D Deficiency Causes Sensorineural Hearing Loss in Mice and Humans" Genes 15, no. 1: 48. https://doi.org/10.3390/genes15010048
APA StyleKalinousky, A. J., Luperchio, T. R., Schrode, K. M., Harris, J. R., Zhang, L., DeLeon, V. B., Fahrner, J. A., Lauer, A. M., & Bjornsson, H. T. (2024). KMT2D Deficiency Causes Sensorineural Hearing Loss in Mice and Humans. Genes, 15(1), 48. https://doi.org/10.3390/genes15010048